Selection of Entries from Clusters/Groups on the basis of Genetic Distances
Source:R/select.distance.R
select.distance.RdSelect entries from cluster/groups in the entire collection by genetic distance based sampling according to allocation specified.
Usage
select.distance(
data,
names,
group,
alloc,
dist.mat,
always.selected = NULL,
method = c("mean.medoid", "median.medoid", "nearest.centroid", "nearest.median",
"mean.peripheral", "median.peripheral", "eccentricity", "farness.centrality",
"kennard.stone", "duplex", "honigs", "farthest.point", "nearest.neighbour", "naes",
"optim.medoid", "hclust.random", "hclust.medoid"),
hclust.method = c("average", "single", "complete", "ward.D", "mcquitty", "median",
"centroid", "ward.D2")
)Arguments
- data
The data as a data frame object. The data frame should possess one row per individual and columns with the individual names and multiple trait/character data.
- names
Name of column with the accession names as a character string.
- group
Name of column with the accession group/cluster names as a character string.
- alloc
A named numeric vector specifying the number of entries to be selected. Names should correspond to the levels of the "
"group"column, and values indicate the number of elements to be selected from each level.- dist.mat
A precomputed distance matrix of distance measures between the accessions in
data.- always.selected
Names of accessions to be always included in the core set as a character vector.
- method
The method for sampling accessions from each cluster/group. Either
"mean.medoid","median.medoid","nearest.centroid","nearest.median","mean.peripheral","median.peripheral","eccentricity","farness.centrality","kennard.stone","duplex","honigs","farthest.point","nearest.neighbour","naes","optim.medoid","hclust.random"or"hclust.medoid". See Methods.- hclust.method
The hierarchical clustering method to be used. Either
"ward.D","ward.D2","single","complete","average"(= UPGMA),"mcquitty"(= WPGMA),"median"(= WPGMC) or"centroid"(= UPGMC).
Details
For each cluster/group, entries are selected by several methods from within-cluster/group genetic distances between accessions according to the allocation provided (See Methods).
Entries listed as always.selected are mandatorily included in the
selection. Warnings are issued if requested allocation is smaller than the
number of always-selected entries in a cluster/group and/or when the
cluster/group does not contain enough remaining entries to fulfill the
allocation.
Methods
Centrality Based Methods
Selects accessions that are most representative/closest to the cluster/group center.
Medoid-like Representative Sampling by Minimal Mean Distance
Selects medoid-like representatives as accessions with the smallest average distance to all others within the group (Kaufman and Rousseeuw 1987; Kaufman and Rousseeuw 1990) .
For each accession \(g\), the mean distance to all other accessions \(h\) is computed as:
\[\bar{d}_g = \frac{1}{G} \sum_{h=1}^{G} d_{gh}\]
Accessions are ranked by \(\bar{d}_g\) in ascending order and the top \(n\) are selected.
Medoid-like Representative Sampling by Minimal Median Distance
Selects medoid-like representatives as accessions with the smallest median distance to all others within the group. This method is less influenced by outliers (Kaufman and Rousseeuw 1987; Kaufman and Rousseeuw 1990) .
For each accession \(g\), the median distance to all other accessions \(h\) is computed as:
\[\tilde{d}_g = \text{median}_{h=1,\dots,G}(d_{gh})\]
Accessions are ranked by \(\tilde{d}_g\) in ascending order and the top \(n\) are selected.
Representative Sampling by Proximity to Group Centroid
Selects accessions closest to the group centroid in principal
coordinate space, computed via multivariate dispersion analysis using
betadisper
(Anderson 2006; Anderson et al. 2006)
.
The distance of each accession \(g\) to the group centroid \(C\) in PCoA space is:
\[\delta_g = \| \mathbf{p}_g - \mathbf{c} \|\]
Where \(\mathbf{p}_g\) is the PCoA coordinate vector of accession \(g\) and \(\mathbf{c}\) is the group centroid. Accessions are ranked by \(\delta_g\) in ascending order and the top \(n\) are selected.
Representative Sampling by Proximity to Group Spatial Median
Selects accessions closest to the group spatial median in principal
coordinate space, computed via multivariate dispersion analysis using
betadisper
(O'Neill and Mathews 2000)
.
The distance of each accession \(g\) to the group spatial median \(M\) is:
\[\delta_g^* = \| \mathbf{p}_g - \mathbf{m} \|\]
where \(\mathbf{m}\) is the spatial median of the group in PCoA space. Accessions are ranked by \(\delta_g^*\) in ascending order and the top \(n\) are selected.
Peripheral/Extremity Based Methods
Selects accessions that are most dissimilar from the rest in a cluster/group i.e. the accessions which are in the boundary or outliers.
Peripheral Sampling by Maximal Mean Distance
Selects the most peripheral accessions as those with the largest average distance to all others within the group (Kaufman and Rousseeuw 1987; Kaufman and Rousseeuw 1990) .
\[\bar{d}_g = \frac{1}{G} \sum_{h=1}^{G} d_{gh}\]
Accessions are ranked by \(\bar{d}_g\) in descending order and the top \(n\) are selected.
Peripheral Sampling by Maximal Median Distance
Selects the most peripheral accessions as those with the largest median distance to all others within the group (Kaufman and Rousseeuw 1987; Kaufman and Rousseeuw 1990) .
\[\tilde{d}_g = \text{median}_{h=1,\dots,G}(d_{gh})\]
Accessions are ranked by \(\tilde{d}_g\) in descending order and the top \(n\) are selected.
Peripheral Sampling by Maximal Eccentricity
Selects accessions with the largest eccentricity — the maximum distance to any other accession in the group (Hage and Harary 1995) .
\[e_g = \max_{h=1,\dots,G} d_{gh}\]
Accessions are ranked by \(e_g\) in descending order and the top \(n\) are selected. Eccentricity captures the worst-case dissimilarity of an accession rather than its average behaviour.
Peripheral Sampling by Maximal Farness Centrality
Selects accessions with the greatest total distance to all others, i.e. those most remote from the rest of the group (Sabidussi 1966) .
\[f_g = \sum_{h=1}^{G} d_{gh}\]
Accessions are ranked by \(f_g\) in descending order and the top
\(n\) are selected. Farness centrality is proportional to
\(\bar{d}_g\) and differs from mean.peripheral only in that it
uses the raw sum rather than the mean, producing identical rankings.
Space-Filling/Coverage Methods
Select accessions that are spread maximally across the feature space in a cluster/group i.e. diversity sampling.
Space-Filling Sampling via the Kennard-Stone Algorithm
Selects \(n\) accessions that maximally and uniformly
cover the distance space via the Kennard-Stone algorithm
(Kennard and Stone 1969)
(See
kenStone).
Starting from the pair of accessions with the largest pairwise distance:
\[\lbrace g_1, g_2 \rbrace = \underset{g,h}{\arg\max}\, d_{gh}\]
each subsequent accession \(g_k\) is selected by maximising its minimum distance to the already-selected set \(S\):
\[g_k = \underset{g \notin S}{\arg\max} \min_{s \in S} d_{gs}\]
This greedy procedure ensures even space coverage without relying on cluster structure.
Space-Filling Sampling via the DUPLEX Algorithm
Extends the Kennard-Stone algorithm to simultaneously construct a model set and a test set with similar distributions (Kennard and Stone 1969; Snee 1977) (duplex). Accessions are selected using Mahalanobis distance:
\[d_M(g, h) = \sqrt{(\mathbf{x}_g - \mathbf{x}_h)^\top \Sigma^{-1} (\mathbf{x}_g - \mathbf{x}_h)}\]
where \(\Sigma\) is the covariance matrix. At each step, the pair maximising \(d_M\) is split alternately between model and test sets, ensuring both sets span the full feature space.
Space-Filling Sampling via the Honigs Algorithm
Selects \(n\) accessions sequentially by maximising dissimilarity to the already-selected set (Honigs et al. 1985) (honigs)
At each step \(k\), the accession \(g_k\) maximising total distance to all previously selected accessions \(S\) is chosen:
\[g_k = \underset{g \notin S}{\arg\max} \sum_{s \in S} d_{gs}\]
This favours accessions that are collectively most dissimilar to the current selection, producing broad coverage of the distance space.
.
Space-Filling Sampling via Farthest-Point (Max-Min) Algorithm
Selects \(n\) accessions by iteratively maximising the minimum distance to the current selected set — also known as the max-min or farthest-point sampling algorithm (Gonzalez 1985; Dyer and Frieze 1985; Hochbaum and Shmoys 1985) .
\[g_k = \underset{g \notin S}{\arg\max} \min_{s \in S} d_{gs}\]
This is equivalent to Kennard-Stone but without the symmetric initialisation step. It provides a deterministic, greedy approximation to the \(k\)-centre problem:
\[\min_{S \subset G,\, |S|=n} \max_{g \in G} \min_{s \in S} d_{gs}\]
Density Based Methods
Select points based on local neighbourhood density.
Density-Based Sampling by Minimal Nearest-Neighbour Distance
Selects accessions residing in the densest regions of the distance space, identified as those with the smallest nearest-neighbour distance (Cover and Hart 1967; Fix and Hodges 1989) .
For each accession \(g\), the nearest-neighbour distance is:
\[\text{nn}_g = \min_{h \neq g} d_{gh}\]
Accessions are ranked by \(\text{nn}_g\) in ascending order and the top \(n\) are selected. Small \(\text{nn}_g\) indicates that \(g\) resides in a dense cluster; this method preferentially samples from high-density regions.
Cluster Based Methods
These methods partition the cluster/group space into sub-clusters/groups, then samples from each one.
Globally Optimal Medoid Sampling via Partitioning Around Medoids (PAM)
Selects a set of \(n\) medoids that jointly minimise the total
distance of every accession to its nearest medoid, via
pam.
The objective function minimised is:
\[\min_{S \subset G,\, |S|=n} \sum_{g=1}^{G} \min_{s \in S} d_{gs}\]
Unlike "mean.medoid", medoids are co-optimised as a set, ensuring
they collectively represent the full distribution of the group rather than
independently scoring each accession.
Cluster-Based Sampling via K-means (Naes Method)
Partitions
accessions into \(n\) clusters via k-means applied to the distance
matrix (See naes), then selects the accession
closest to each cluster centre as the representative
(Naes 1987; Naes et al. 2017)
.
The k-means objective minimised is:
\[\min \sum_{k=1}^{n} \sum_{g \in C_k} d_{g, \mu_k}^2\]
where \(C_k\) is the \(k\)-th cluster and \(\mu_k\) is its centre. One representative per cluster is returned, ensuring broad, partition-aware coverage.
Cluster-Based Sampling via Hierarchical Clustering with Random Selection
Partitions accessions into \(n\) clusters by cutting a hierarchical clustering dendrogram at height \(k = n\), then randomly samples one accession from each cluster (Ward 1963; Li et al. 2002) .
The dendrogram is built by agglomerative hierarchical clustering using the
linkage criterion specified by hclust. For
clusters \(C_1, \dots, C_n\), one accession is drawn uniformly at
random from each:
\[g_k \sim \text{Uniform}(C_k), \quad k = 1, \dots, n\]
This introduces stochasticity within a structured partition, balancing coverage with randomness.
Cluster-Based Sampling via Hierarchical Clustering with Medoid Selection
Partitions accessions into \(n\) clusters by cutting a hierarchical clustering dendrogram at height \(k = n\), then selects the within-cluster medoid as the representative of each cluster (Kaufman and Rousseeuw 1987; Ward 1963) .
For each cluster \(C_k\), the medoid is the accession minimising total within-cluster distance:
\[g_k^* = \underset{g \in C_k}{\arg\min} \sum_{h \in C_k} d_{gh}\]
This combines the structured partitioning of hierarchical clustering with deterministic, centrality-based representative selection.
References
Anderson MJ (2006).
“Distance-based tests for homogeneity of multivariate dispersions.”
Biometrics, 62(1), 245–253.
Anderson MJ, Ellingsen KE, McArdle BH (2006).
“Multivariate dispersion as a measure of beta diversity.”
Ecology Letters, 9(6), 683–693.
Cover T, Hart P (1967).
“Nearest neighbor pattern classification.”
IEEE Transactions on Information Theory, 13(1), 21–27.
Dyer ME, Frieze AM (1985).
“A simple heuristic for the p-centre problem.”
Operations Research Letters, 3(6), 285–288.
Fix E, Hodges JL (1989).
“Discriminatory analysis - Nonparametric discrimination: Consistency properties.”
International Statistical Review / Revue Internationale de Statistique, 57(3), 238–247.
Gonzalez TF (1985).
“Clustering to minimize the maximum intercluster distance.”
Theoretical Computer Science, 38, 293–306.
Hage P, Harary F (1995).
“Eccentricity and centrality in networks.”
Social Networks, 17(1), 57–63.
Hochbaum DS, Shmoys DB (1985).
“A best possible heuristic for the K-center problem.”
Mathematics of Operations Research, 10(2), 180–184.
Honigs DE, Hieftje GM, Mark HL, Hirschfeld TB (1985).
“Unique-sample selection via near-infrared spectral subtraction.”
Analytical Chemistry, 57(12), 2299–2303.
Kaufman L, Rousseeuw PJ (1990).
Finding Groups in Data: An Introduction to Cluster Analysis, Wiley Series in Probability and Statistics, 1 edition.
Wiley.
ISBN 978-0-471-87876-6 978-0-470-31680-1.
Kaufman P, Rousseeuw PJ (1987).
“Clustering by means of medoids.”
In Dodge Y (ed.), Proceedings of the Statistical Data Analysis Based on the L1 Norm Conference, Neuchatel, Switzerland, volume 31, 405–416.
Kennard RW, Stone LA (1969).
“Computer aided design of experiments.”
Technometrics, 11(1), 137–148.
Li Z, Zhang H, Zeng Y, Yang Z, Shen S, Sun C, Wang X (2002).
“Studies on sampling schemes for the establishment of core collection of rice landraces in Yunnan, China.”
Genetic Resources and Crop Evolution, 49(1), 67–74.
Naes T (1987).
“The design of calibration in near infra-red reflectance analysis by clustering.”
Journal of Chemometrics, 1(2), 121–134.
Naes T, Isaksson T, Fearn T, Davies T (2017).
A User-Friendly Guide to Multivariate Calibration and Classification, Second edition edition.
IM Publications LLP, Chichester.
ISBN 978-1-906715-25-0.
O'Neill ME, Mathews K (2000).
“A weighted least squares approach to levene's test of homogeneity of variance.”
Australian & New Zealand Journal of Statistics, 42(1), 81–100.
Sabidussi G (1966).
“The centrality index of a graph.”
Psychometrika, 31(4), 581–603.
Snee RD (1977).
“Validation of regression models: Methods and examples.”
Technometrics, 19(4), 415–428.
Ward JH (1963).
“Hierarchical grouping to optimize an objective function.”
Journal of the American Statistical Association, 58(301), 236–244.
Examples
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Prepare example data
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
library(cluster)
library(ggplot2)
data(cassava_EC_gp)
set.seed(123)
data <- cassava_EC_gp[sample(1:nrow(cassava_EC_gp), 500), ]
quant <- c("NMSR", "TTRN", "TFWSR", "TTRW", "TFWSS", "TTSW", "TTPW", "AVPW",
"ARSR", "SRDM")
qual <- c("CUAL", "LNGS", "PTLC", "DSTA", "LFRT", "LBTEF", "CBTR", "NMLB",
"ANGB", "CUAL9M", "LVC9M", "TNPR9M", "PL9M", "STRP", "STRC",
"PSTR")
data[, qual] <- lapply(data[, qual], as.factor)
# Get the Gower's distance matrix
dist_matrix <- daisy(x = data[, c(qual, quant)],
metric = "gower")
data <- cbind(genotypes = rownames(data), data)
row.names(data) <- NULL
# Prepare inputs
counts <- c(I = 16, II = 15, III = 9, IV = 18, V = 20, VI = 8)
mand_accns <-
c("TMe-2018", "TMe-801", "TMe-3191", "TMe-1830", "TMe-1790")
gp_vec <- setNames(as.character(data[, "Cluster"]), data[, "genotypes"])
# \donttest{
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Fetch selected accessions by centrality based methods
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Medoid-like Representative Sampling by Minimal Mean Distance
sel_mean_medoid_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "mean.medoid")
sel_mean_medoid_out
#> $I
#> [1] "TMe-1823" "TMe-2066" "TMe-1717" "TMe-2152" "TMe-1914" "TMe-910"
#> [7] "TMe-3623" "TMe-1451" "TMe-2964" "TMe-469" "TMe-3104" "TMe-882"
#> [13] "TMe-952" "TMe-1930" "TMe-2103" "TMe-1830"
#>
#> $II
#> [1] "TMe-3495" "TMe-455" "TMe-2951" "TMe-890" "TMe-3239" "TMe-2715"
#> [7] "TMe-681" "TMe-2258" "TMe-409" "TMe-1698" "TMe-1323" "TMe-2997"
#> [13] "TMe-960" "TMe-1474" "TMe-796"
#>
#> $III
#> [1] "TMe-1725" "TMe-3335" "TMe-2748" "TMe-2169" "TMe-617" "TMe-3207" "TMe-3336"
#> [8] "TMe-2356" "TMe-1790"
#>
#> $IV
#> [1] "TMe-428" "TMe-1996" "TMe-1765" "TMe-2367" "TMe-3327" "TMe-3214"
#> [7] "TMe-3232" "TMe-2375" "TMe-3218" "TMe-1139" "TMe-1167" "TMe-3378"
#> [13] "TMe-840" "TMe-65" "TMe-3108" "TMe-2039" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-332" "TMe-574" "TMe-1414" "TMe-1979" "TMe-363" "TMe-1160"
#> [7] "TMe-1268" "TMe-1622" "TMe-312" "TMe-1234" "TMe-2271" "TMe-1559"
#> [13] "TMe-360" "TMe-1299" "TMe-245" "TMe-745" "TMe-645" "TMe-647"
#> [19] "TMe-2441" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-936" "TMe-222" "TMe-1481" "TMe-505" "TMe-1676" "TMe-1592"
#> [8] "TMe-1217"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_mean_medoid_out,
use.names = FALSE)) +
labs(title = "mean.medoid")
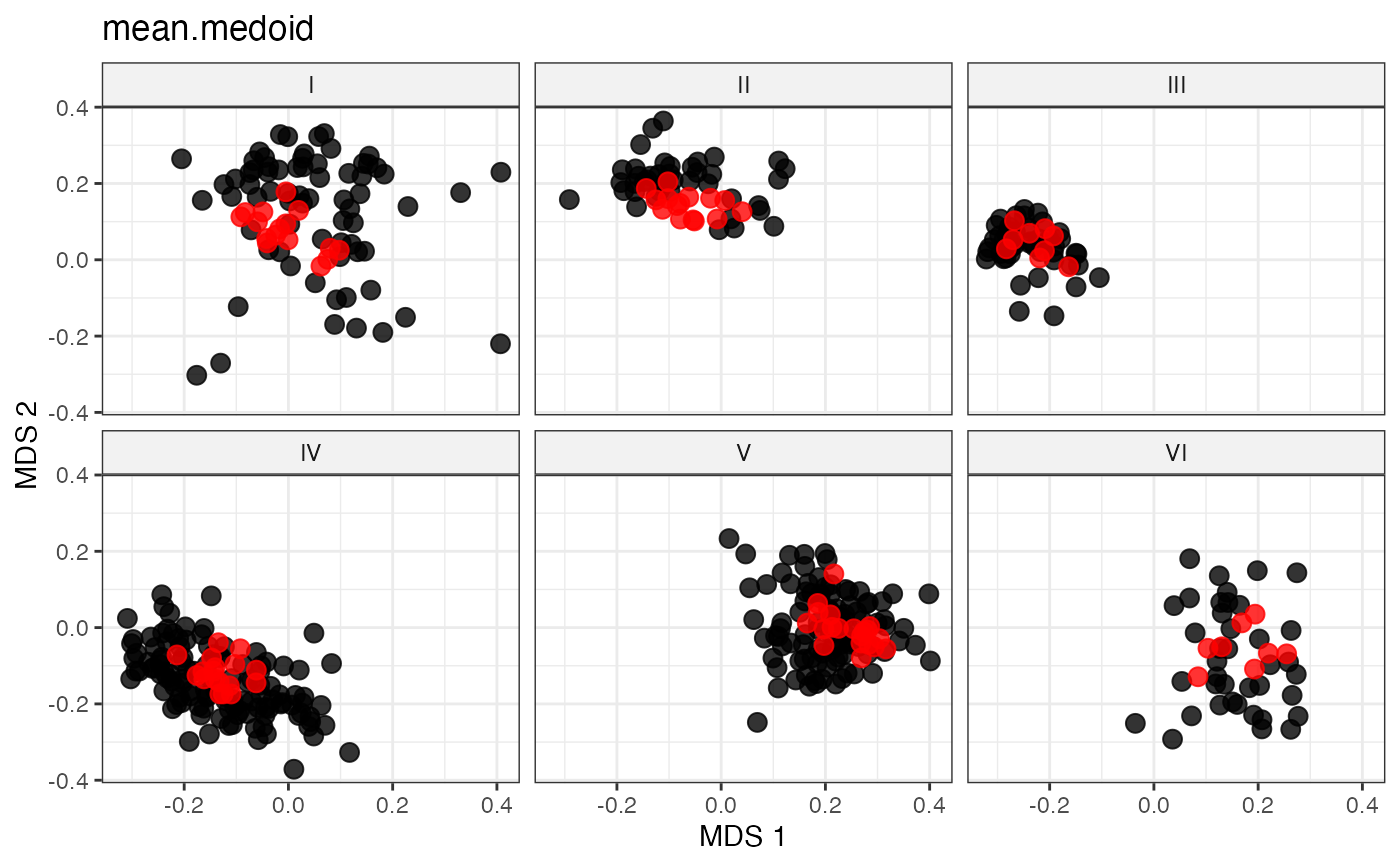 # Medoid-like Representative Sampling by Minimal Median Distance
sel_median_medoid_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "median.medoid")
sel_median_medoid_out
#> $I
#> [1] "TMe-1823" "TMe-2066" "TMe-1914" "TMe-469" "TMe-1451" "TMe-2152"
#> [7] "TMe-3142" "TMe-3623" "TMe-910" "TMe-1717" "TMe-952" "TMe-3548"
#> [13] "TMe-1117" "TMe-882" "TMe-3719" "TMe-1830"
#>
#> $II
#> [1] "TMe-455" "TMe-3495" "TMe-2951" "TMe-3239" "TMe-890" "TMe-2715"
#> [7] "TMe-960" "TMe-2997" "TMe-681" "TMe-409" "TMe-1831" "TMe-1698"
#> [13] "TMe-2258" "TMe-1474" "TMe-2611"
#>
#> $III
#> [1] "TMe-1725" "TMe-2169" "TMe-3207" "TMe-3335" "TMe-2270" "TMe-617" "TMe-2748"
#> [8] "TMe-64" "TMe-1790"
#>
#> $IV
#> [1] "TMe-1765" "TMe-1996" "TMe-428" "TMe-840" "TMe-3108" "TMe-3218"
#> [7] "TMe-2367" "TMe-1167" "TMe-3327" "TMe-2375" "TMe-3072" "TMe-3257"
#> [13] "TMe-3378" "TMe-3562" "TMe-3602" "TMe-3232" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-332" "TMe-1414" "TMe-363" "TMe-1268" "TMe-574" "TMe-312"
#> [7] "TMe-1160" "TMe-1979" "TMe-1234" "TMe-745" "TMe-1299" "TMe-1559"
#> [13] "TMe-2271" "TMe-1622" "TMe-645" "TMe-360" "TMe-543" "TMe-1273"
#> [19] "TMe-647" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-222" "TMe-936" "TMe-1481" "TMe-1676" "TMe-505" "TMe-1592"
#> [8] "TMe-1945"
#>
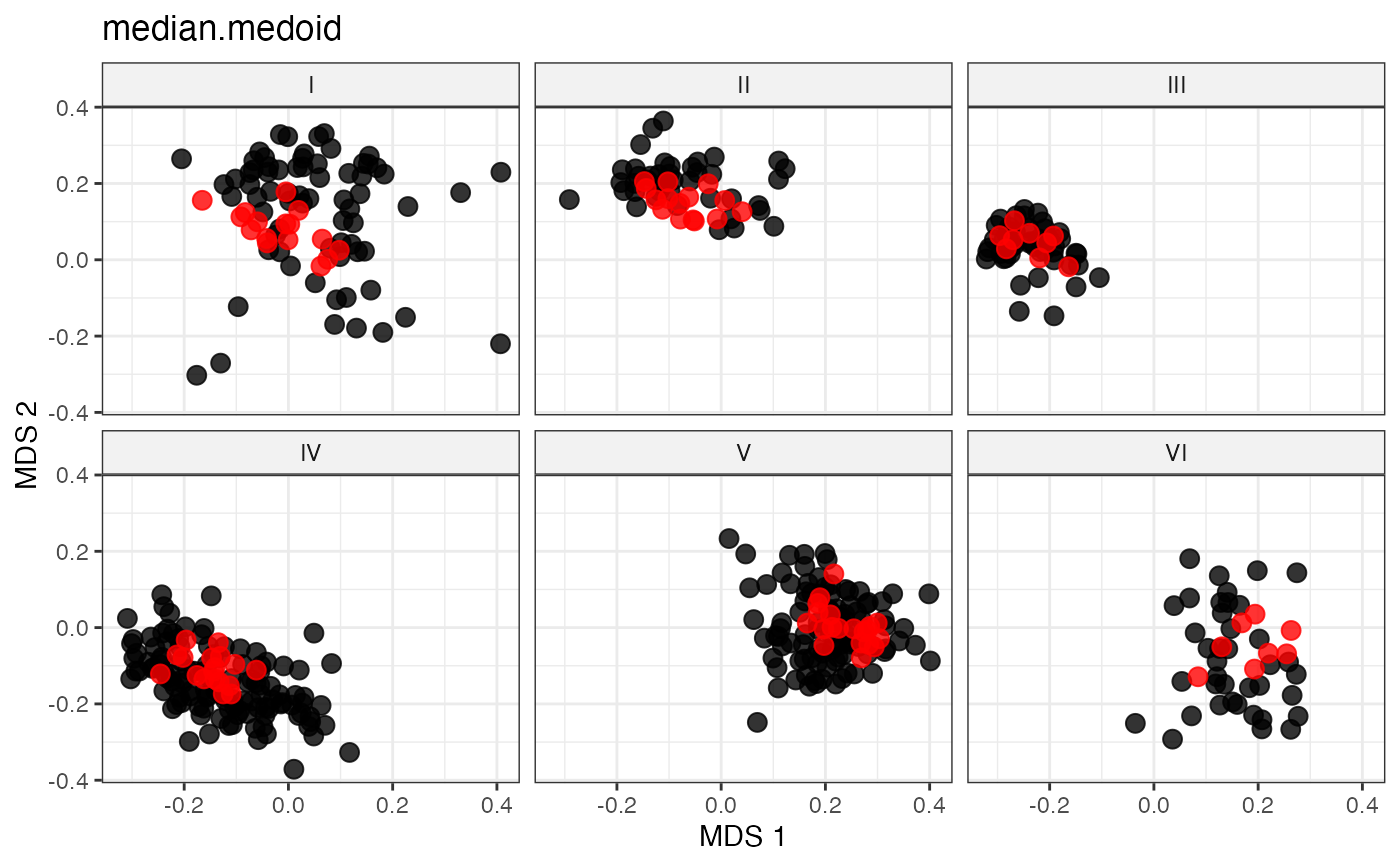
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_median_medoid_out,
use.names = FALSE)) +
labs(title = "median.medoid")
# Medoid-like Representative Sampling by Minimal Median Distance
sel_median_medoid_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "median.medoid")
sel_median_medoid_out
#> $I
#> [1] "TMe-1823" "TMe-2066" "TMe-1914" "TMe-469" "TMe-1451" "TMe-2152"
#> [7] "TMe-3142" "TMe-3623" "TMe-910" "TMe-1717" "TMe-952" "TMe-3548"
#> [13] "TMe-1117" "TMe-882" "TMe-3719" "TMe-1830"
#>
#> $II
#> [1] "TMe-455" "TMe-3495" "TMe-2951" "TMe-3239" "TMe-890" "TMe-2715"
#> [7] "TMe-960" "TMe-2997" "TMe-681" "TMe-409" "TMe-1831" "TMe-1698"
#> [13] "TMe-2258" "TMe-1474" "TMe-2611"
#>
#> $III
#> [1] "TMe-1725" "TMe-2169" "TMe-3207" "TMe-3335" "TMe-2270" "TMe-617" "TMe-2748"
#> [8] "TMe-64" "TMe-1790"
#>
#> $IV
#> [1] "TMe-1765" "TMe-1996" "TMe-428" "TMe-840" "TMe-3108" "TMe-3218"
#> [7] "TMe-2367" "TMe-1167" "TMe-3327" "TMe-2375" "TMe-3072" "TMe-3257"
#> [13] "TMe-3378" "TMe-3562" "TMe-3602" "TMe-3232" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-332" "TMe-1414" "TMe-363" "TMe-1268" "TMe-574" "TMe-312"
#> [7] "TMe-1160" "TMe-1979" "TMe-1234" "TMe-745" "TMe-1299" "TMe-1559"
#> [13] "TMe-2271" "TMe-1622" "TMe-645" "TMe-360" "TMe-543" "TMe-1273"
#> [19] "TMe-647" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-222" "TMe-936" "TMe-1481" "TMe-1676" "TMe-505" "TMe-1592"
#> [8] "TMe-1945"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_median_medoid_out,
use.names = FALSE)) +
labs(title = "median.medoid")
 # Representative Sampling by Proximity to Group Centroid
sel_group_centroid_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "nearest.centroid")
sel_group_centroid_out
#> $I
#> [1] "TMe-1823" "TMe-2066" "TMe-1717" "TMe-3351" "TMe-1914" "TMe-2152"
#> [7] "TMe-3623" "TMe-1451" "TMe-469" "TMe-1830" "TMe-952" "TMe-1096"
#> [13] "TMe-2103" "TMe-3437" "TMe-1930" "TMe-1830"
#>
#> $II
#> [1] "TMe-3495" "TMe-455" "TMe-2951" "TMe-890" "TMe-3239" "TMe-2715"
#> [7] "TMe-681" "TMe-2258" "TMe-409" "TMe-1698" "TMe-1323" "TMe-2997"
#> [13] "TMe-960" "TMe-1474" "TMe-796"
#>
#> $III
#> [1] "TMe-1725" "TMe-3335" "TMe-2169" "TMe-2748" "TMe-617" "TMe-3207" "TMe-2356"
#> [8] "TMe-3336" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2755" "TMe-3196" "TMe-3591" "TMe-3269" "TMe-1765" "TMe-2841"
#> [7] "TMe-2567" "TMe-1016" "TMe-3390" "TMe-2928" "TMe-2375" "TMe-3068"
#> [13] "TMe-3409" "TMe-368" "TMe-186" "TMe-1377" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1293" "TMe-574" "TMe-1453" "TMe-1101" "TMe-1500" "TMe-2753"
#> [7] "TMe-1880" "TMe-1003" "TMe-647" "TMe-1788" "TMe-755" "TMe-423"
#> [13] "TMe-2057" "TMe-2271" "TMe-1299" "TMe-532" "TMe-1427" "TMe-323"
#> [19] "TMe-877" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-936" "TMe-222" "TMe-1481" "TMe-505" "TMe-1676" "TMe-1592"
#> [8] "TMe-1217"
#>
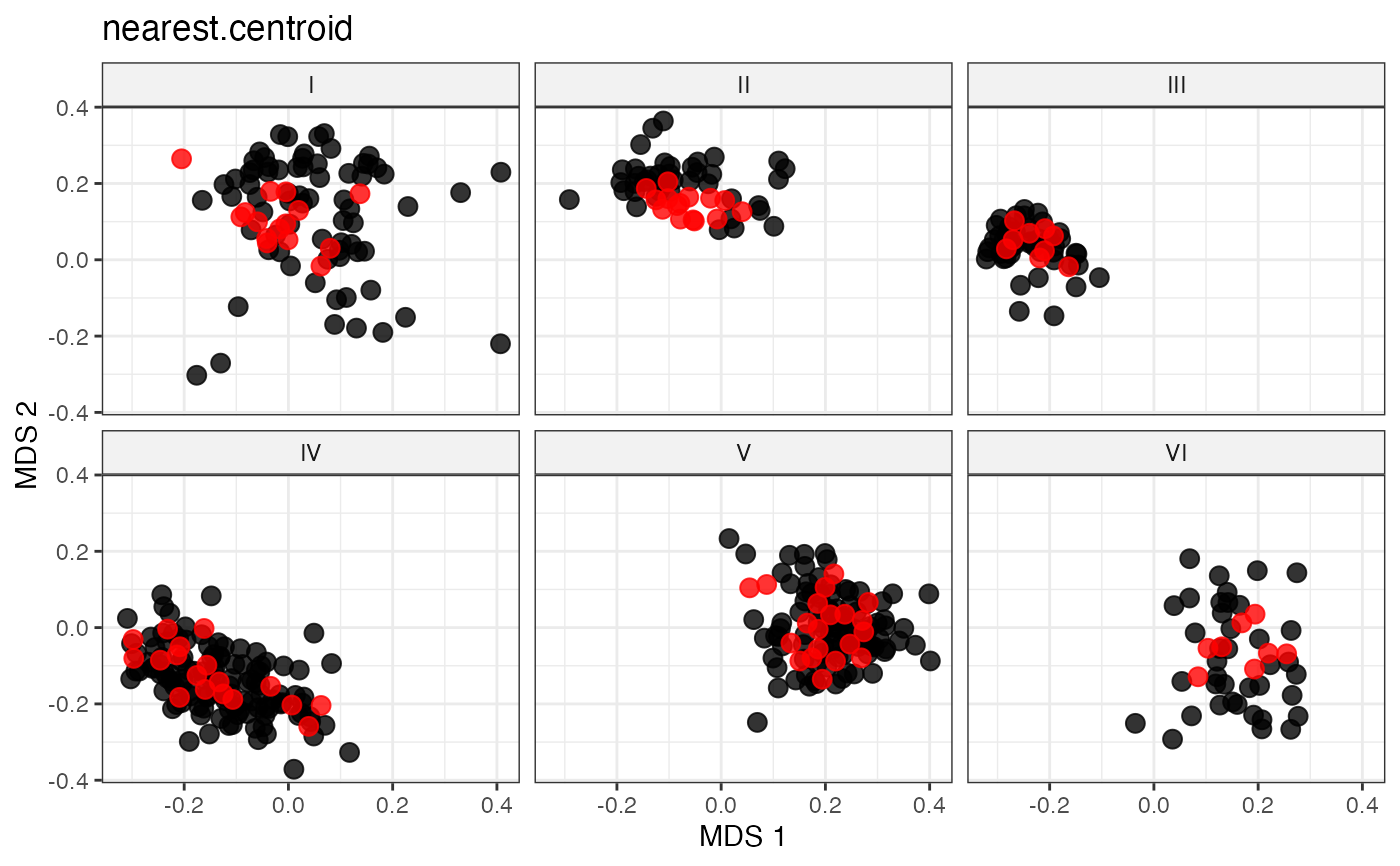
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_group_centroid_out,
use.names = FALSE)) +
labs(title = "nearest.centroid")
# Representative Sampling by Proximity to Group Centroid
sel_group_centroid_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "nearest.centroid")
sel_group_centroid_out
#> $I
#> [1] "TMe-1823" "TMe-2066" "TMe-1717" "TMe-3351" "TMe-1914" "TMe-2152"
#> [7] "TMe-3623" "TMe-1451" "TMe-469" "TMe-1830" "TMe-952" "TMe-1096"
#> [13] "TMe-2103" "TMe-3437" "TMe-1930" "TMe-1830"
#>
#> $II
#> [1] "TMe-3495" "TMe-455" "TMe-2951" "TMe-890" "TMe-3239" "TMe-2715"
#> [7] "TMe-681" "TMe-2258" "TMe-409" "TMe-1698" "TMe-1323" "TMe-2997"
#> [13] "TMe-960" "TMe-1474" "TMe-796"
#>
#> $III
#> [1] "TMe-1725" "TMe-3335" "TMe-2169" "TMe-2748" "TMe-617" "TMe-3207" "TMe-2356"
#> [8] "TMe-3336" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2755" "TMe-3196" "TMe-3591" "TMe-3269" "TMe-1765" "TMe-2841"
#> [7] "TMe-2567" "TMe-1016" "TMe-3390" "TMe-2928" "TMe-2375" "TMe-3068"
#> [13] "TMe-3409" "TMe-368" "TMe-186" "TMe-1377" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1293" "TMe-574" "TMe-1453" "TMe-1101" "TMe-1500" "TMe-2753"
#> [7] "TMe-1880" "TMe-1003" "TMe-647" "TMe-1788" "TMe-755" "TMe-423"
#> [13] "TMe-2057" "TMe-2271" "TMe-1299" "TMe-532" "TMe-1427" "TMe-323"
#> [19] "TMe-877" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-936" "TMe-222" "TMe-1481" "TMe-505" "TMe-1676" "TMe-1592"
#> [8] "TMe-1217"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_group_centroid_out,
use.names = FALSE)) +
labs(title = "nearest.centroid")
 # Representative Sampling by Proximity to Group Spatial Median
sel_group_median_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "nearest.median")
sel_group_median_out
#> $I
#> [1] "TMe-1823" "TMe-2066" "TMe-1717" "TMe-3351" "TMe-1914" "TMe-2152"
#> [7] "TMe-1451" "TMe-3623" "TMe-469" "TMe-1830" "TMe-952" "TMe-2103"
#> [13] "TMe-1096" "TMe-1930" "TMe-3437" "TMe-1830"
#>
#> $II
#> [1] "TMe-3495" "TMe-455" "TMe-2951" "TMe-890" "TMe-3239" "TMe-2715"
#> [7] "TMe-681" "TMe-2258" "TMe-409" "TMe-1698" "TMe-1323" "TMe-2997"
#> [13] "TMe-1474" "TMe-960" "TMe-796"
#>
#> $III
#> [1] "TMe-1725" "TMe-3335" "TMe-2169" "TMe-617" "TMe-2748" "TMe-3207" "TMe-2356"
#> [8] "TMe-3336" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2755" "TMe-3196" "TMe-3591" "TMe-1765" "TMe-3269" "TMe-2567"
#> [7] "TMe-1016" "TMe-3390" "TMe-2841" "TMe-3068" "TMe-2375" "TMe-2928"
#> [13] "TMe-186" "TMe-3409" "TMe-368" "TMe-1150" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1293" "TMe-1453" "TMe-574" "TMe-1500" "TMe-1101" "TMe-2753"
#> [7] "TMe-1003" "TMe-647" "TMe-1880" "TMe-1788" "TMe-755" "TMe-2057"
#> [13] "TMe-423" "TMe-2271" "TMe-532" "TMe-1299" "TMe-1427" "TMe-877"
#> [19] "TMe-323" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-936" "TMe-222" "TMe-1481" "TMe-505" "TMe-1676" "TMe-1592"
#> [8] "TMe-1945"
#>
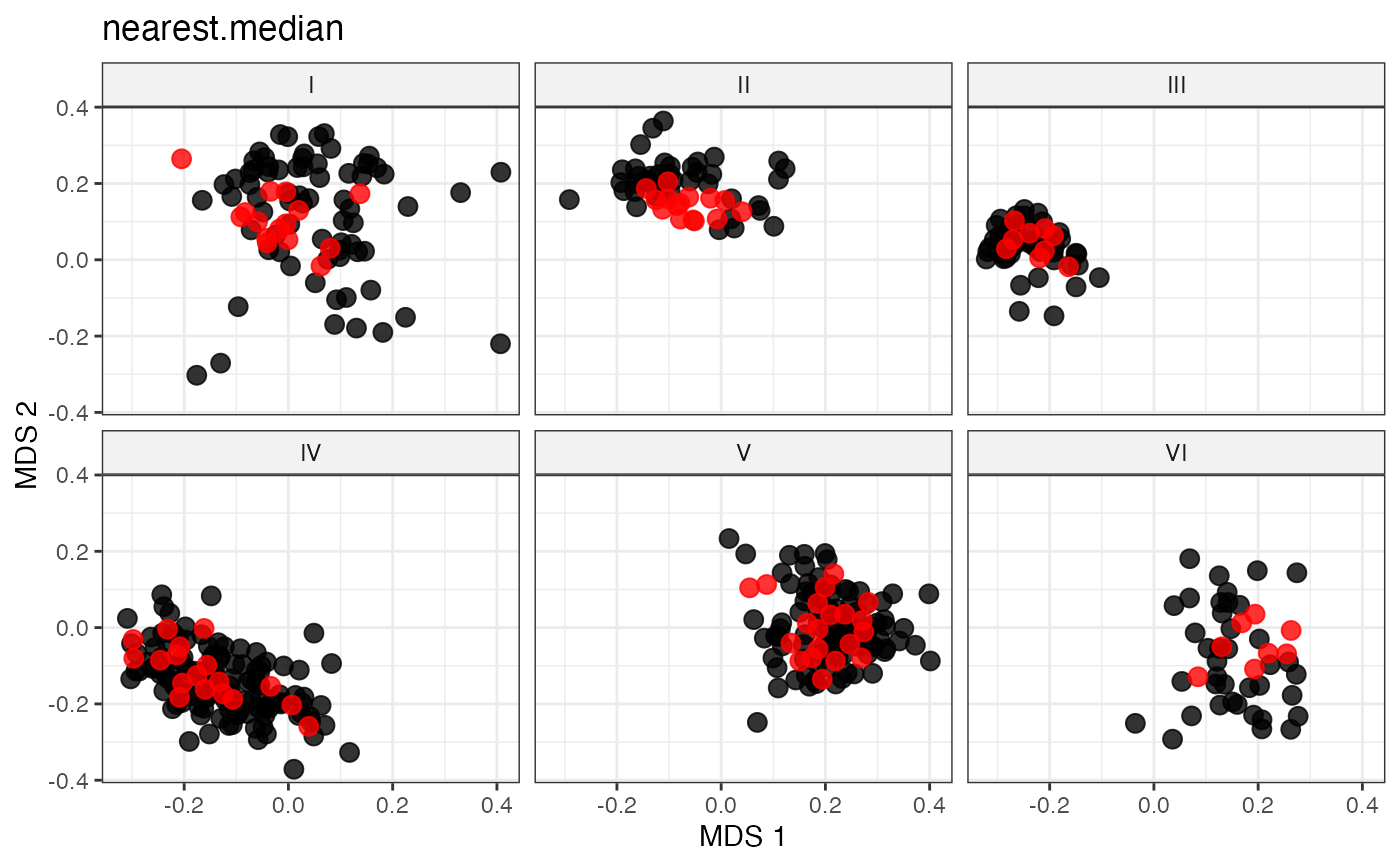
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_group_median_out,
use.names = FALSE)) +
labs(title = "nearest.median")
# Representative Sampling by Proximity to Group Spatial Median
sel_group_median_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "nearest.median")
sel_group_median_out
#> $I
#> [1] "TMe-1823" "TMe-2066" "TMe-1717" "TMe-3351" "TMe-1914" "TMe-2152"
#> [7] "TMe-1451" "TMe-3623" "TMe-469" "TMe-1830" "TMe-952" "TMe-2103"
#> [13] "TMe-1096" "TMe-1930" "TMe-3437" "TMe-1830"
#>
#> $II
#> [1] "TMe-3495" "TMe-455" "TMe-2951" "TMe-890" "TMe-3239" "TMe-2715"
#> [7] "TMe-681" "TMe-2258" "TMe-409" "TMe-1698" "TMe-1323" "TMe-2997"
#> [13] "TMe-1474" "TMe-960" "TMe-796"
#>
#> $III
#> [1] "TMe-1725" "TMe-3335" "TMe-2169" "TMe-617" "TMe-2748" "TMe-3207" "TMe-2356"
#> [8] "TMe-3336" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2755" "TMe-3196" "TMe-3591" "TMe-1765" "TMe-3269" "TMe-2567"
#> [7] "TMe-1016" "TMe-3390" "TMe-2841" "TMe-3068" "TMe-2375" "TMe-2928"
#> [13] "TMe-186" "TMe-3409" "TMe-368" "TMe-1150" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1293" "TMe-1453" "TMe-574" "TMe-1500" "TMe-1101" "TMe-2753"
#> [7] "TMe-1003" "TMe-647" "TMe-1880" "TMe-1788" "TMe-755" "TMe-2057"
#> [13] "TMe-423" "TMe-2271" "TMe-532" "TMe-1299" "TMe-1427" "TMe-877"
#> [19] "TMe-323" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-936" "TMe-222" "TMe-1481" "TMe-505" "TMe-1676" "TMe-1592"
#> [8] "TMe-1945"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_group_median_out,
use.names = FALSE)) +
labs(title = "nearest.median")
 #~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Fetch selected accessions by peripheral/extremity based methods
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Peripheral Sampling by Maximal Mean Distance
sel_mean_peripheral_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "mean.peripheral")
sel_mean_peripheral_out
#> $I
#> [1] "TMe-2967" "TMe-3685" "TMe-1425" "TMe-2943" "TMe-41" "TMe-3437"
#> [7] "TMe-3115" "TMe-3398" "TMe-500" "TMe-1190" "TMe-815" "TMe-569"
#> [13] "TMe-3726" "TMe-2027" "TMe-2934" "TMe-1830"
#>
#> $II
#> [1] "TMe-3766" "TMe-2952" "TMe-2033" "TMe-369" "TMe-3800" "TMe-3200"
#> [7] "TMe-674" "TMe-2329" "TMe-1385" "TMe-171" "TMe-2352" "TMe-2568"
#> [13] "TMe-3805" "TMe-1754" "TMe-3101"
#>
#> $III
#> [1] "TMe-3631" "TMe-234" "TMe-261" "TMe-425" "TMe-70" "TMe-381" "TMe-1804"
#> [8] "TMe-2374" "TMe-1790"
#>
#> $IV
#> [1] "TMe-241" "TMe-761" "TMe-812" "TMe-1434" "TMe-2924" "TMe-698"
#> [7] "TMe-1988" "TMe-1020" "TMe-3273" "TMe-2567" "TMe-2956" "TMe-427"
#> [13] "TMe-27" "TMe-3255" "TMe-1376" "TMe-2971" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-419" "TMe-2853" "TMe-2355" "TMe-723" "TMe-712" "TMe-603"
#> [7] "TMe-2907" "TMe-2003" "TMe-344" "TMe-532" "TMe-3329" "TMe-730"
#> [13] "TMe-1159" "TMe-2290" "TMe-2750" "TMe-98" "TMe-1375" "TMe-755"
#> [19] "TMe-1534" "TMe-2018"
#>
#> $VI
#> [1] "TMe-2983" "TMe-1403" "TMe-1124" "TMe-693" "TMe-751" "TMe-1992" "TMe-2791"
#> [8] "TMe-1035"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_mean_peripheral_out,
use.names = FALSE)) +
labs(title = "mean.peripheral")
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Fetch selected accessions by peripheral/extremity based methods
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Peripheral Sampling by Maximal Mean Distance
sel_mean_peripheral_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "mean.peripheral")
sel_mean_peripheral_out
#> $I
#> [1] "TMe-2967" "TMe-3685" "TMe-1425" "TMe-2943" "TMe-41" "TMe-3437"
#> [7] "TMe-3115" "TMe-3398" "TMe-500" "TMe-1190" "TMe-815" "TMe-569"
#> [13] "TMe-3726" "TMe-2027" "TMe-2934" "TMe-1830"
#>
#> $II
#> [1] "TMe-3766" "TMe-2952" "TMe-2033" "TMe-369" "TMe-3800" "TMe-3200"
#> [7] "TMe-674" "TMe-2329" "TMe-1385" "TMe-171" "TMe-2352" "TMe-2568"
#> [13] "TMe-3805" "TMe-1754" "TMe-3101"
#>
#> $III
#> [1] "TMe-3631" "TMe-234" "TMe-261" "TMe-425" "TMe-70" "TMe-381" "TMe-1804"
#> [8] "TMe-2374" "TMe-1790"
#>
#> $IV
#> [1] "TMe-241" "TMe-761" "TMe-812" "TMe-1434" "TMe-2924" "TMe-698"
#> [7] "TMe-1988" "TMe-1020" "TMe-3273" "TMe-2567" "TMe-2956" "TMe-427"
#> [13] "TMe-27" "TMe-3255" "TMe-1376" "TMe-2971" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-419" "TMe-2853" "TMe-2355" "TMe-723" "TMe-712" "TMe-603"
#> [7] "TMe-2907" "TMe-2003" "TMe-344" "TMe-532" "TMe-3329" "TMe-730"
#> [13] "TMe-1159" "TMe-2290" "TMe-2750" "TMe-98" "TMe-1375" "TMe-755"
#> [19] "TMe-1534" "TMe-2018"
#>
#> $VI
#> [1] "TMe-2983" "TMe-1403" "TMe-1124" "TMe-693" "TMe-751" "TMe-1992" "TMe-2791"
#> [8] "TMe-1035"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_mean_peripheral_out,
use.names = FALSE)) +
labs(title = "mean.peripheral")
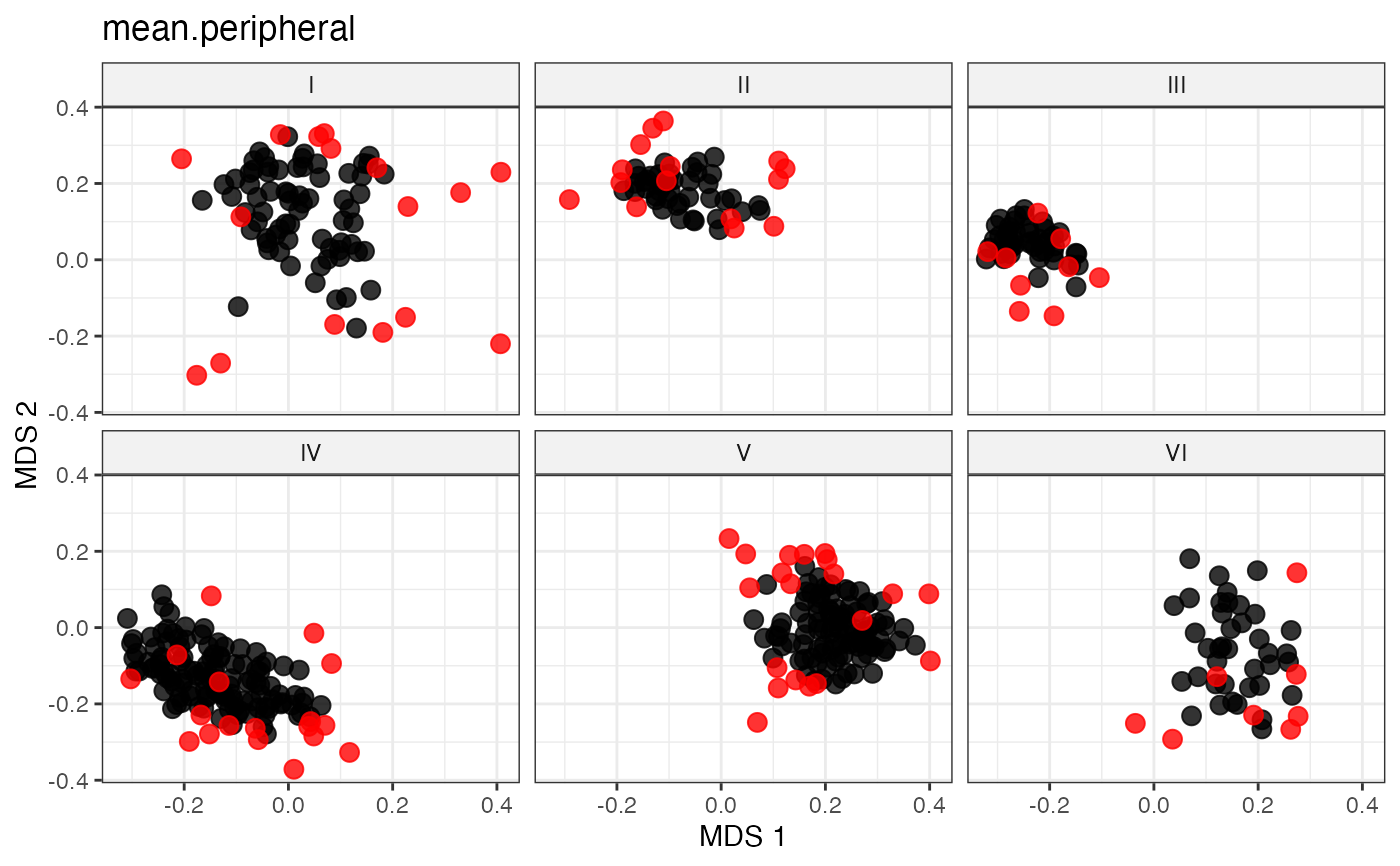 # Peripheral Sampling by Maximal Median Distance
sel_median_peripheral_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "median.peripheral")
sel_median_peripheral_out
#> $I
#> [1] "TMe-3685" "TMe-2967" "TMe-2943" "TMe-1425" "TMe-41" "TMe-3437"
#> [7] "TMe-3115" "TMe-3398" "TMe-1190" "TMe-569" "TMe-2934" "TMe-815"
#> [13] "TMe-3726" "TMe-500" "TMe-3130" "TMe-1830"
#>
#> $II
#> [1] "TMe-369" "TMe-3766" "TMe-3800" "TMe-2033" "TMe-2952" "TMe-674"
#> [7] "TMe-1385" "TMe-3200" "TMe-2329" "TMe-171" "TMe-2568" "TMe-2352"
#> [13] "TMe-3805" "TMe-1754" "TMe-196"
#>
#> $III
#> [1] "TMe-3631" "TMe-234" "TMe-261" "TMe-425" "TMe-1804" "TMe-70" "TMe-381"
#> [8] "TMe-3715" "TMe-1790"
#>
#> $IV
#> [1] "TMe-241" "TMe-1434" "TMe-812" "TMe-761" "TMe-2924" "TMe-698"
#> [7] "TMe-1020" "TMe-1988" "TMe-427" "TMe-3273" "TMe-2567" "TMe-2971"
#> [13] "TMe-2956" "TMe-1376" "TMe-3189" "TMe-3527" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-419" "TMe-2853" "TMe-2355" "TMe-723" "TMe-712" "TMe-2907"
#> [7] "TMe-603" "TMe-344" "TMe-1159" "TMe-3329" "TMe-730" "TMe-98"
#> [13] "TMe-532" "TMe-2003" "TMe-1375" "TMe-2290" "TMe-755" "TMe-1534"
#> [19] "TMe-487" "TMe-2018"
#>
#> $VI
#> [1] "TMe-2983" "TMe-1403" "TMe-1124" "TMe-751" "TMe-1992" "TMe-693" "TMe-2791"
#> [8] "TMe-1035"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_median_peripheral_out,
use.names = FALSE)) +
labs(title = "median.peripheral")
# Peripheral Sampling by Maximal Median Distance
sel_median_peripheral_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "median.peripheral")
sel_median_peripheral_out
#> $I
#> [1] "TMe-3685" "TMe-2967" "TMe-2943" "TMe-1425" "TMe-41" "TMe-3437"
#> [7] "TMe-3115" "TMe-3398" "TMe-1190" "TMe-569" "TMe-2934" "TMe-815"
#> [13] "TMe-3726" "TMe-500" "TMe-3130" "TMe-1830"
#>
#> $II
#> [1] "TMe-369" "TMe-3766" "TMe-3800" "TMe-2033" "TMe-2952" "TMe-674"
#> [7] "TMe-1385" "TMe-3200" "TMe-2329" "TMe-171" "TMe-2568" "TMe-2352"
#> [13] "TMe-3805" "TMe-1754" "TMe-196"
#>
#> $III
#> [1] "TMe-3631" "TMe-234" "TMe-261" "TMe-425" "TMe-1804" "TMe-70" "TMe-381"
#> [8] "TMe-3715" "TMe-1790"
#>
#> $IV
#> [1] "TMe-241" "TMe-1434" "TMe-812" "TMe-761" "TMe-2924" "TMe-698"
#> [7] "TMe-1020" "TMe-1988" "TMe-427" "TMe-3273" "TMe-2567" "TMe-2971"
#> [13] "TMe-2956" "TMe-1376" "TMe-3189" "TMe-3527" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-419" "TMe-2853" "TMe-2355" "TMe-723" "TMe-712" "TMe-2907"
#> [7] "TMe-603" "TMe-344" "TMe-1159" "TMe-3329" "TMe-730" "TMe-98"
#> [13] "TMe-532" "TMe-2003" "TMe-1375" "TMe-2290" "TMe-755" "TMe-1534"
#> [19] "TMe-487" "TMe-2018"
#>
#> $VI
#> [1] "TMe-2983" "TMe-1403" "TMe-1124" "TMe-751" "TMe-1992" "TMe-693" "TMe-2791"
#> [8] "TMe-1035"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_median_peripheral_out,
use.names = FALSE)) +
labs(title = "median.peripheral")
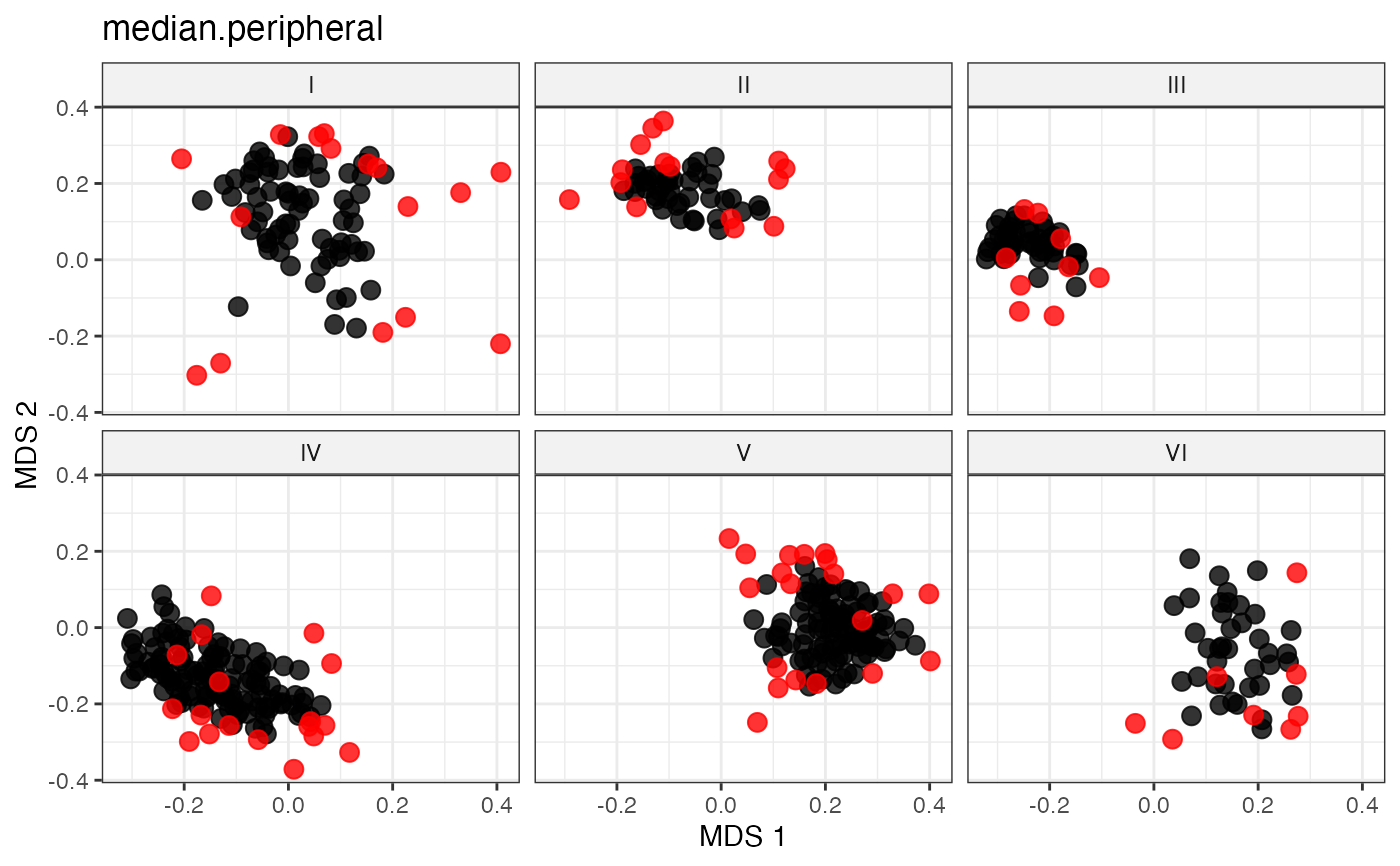 # Peripheral Sampling by Maximal Eccentricity
sel_eccentricity_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "eccentricity")
sel_eccentricity_out
#> $I
#> [1] "TMe-2943" "TMe-3437" "TMe-3719" "TMe-3685" "TMe-3110" "TMe-1532"
#> [7] "TMe-3353" "TMe-1218" "TMe-306" "TMe-2027" "TMe-1117" "TMe-2103"
#> [13] "TMe-2967" "TMe-2810" "TMe-865" "TMe-1830"
#>
#> $II
#> [1] "TMe-2352" "TMe-369" "TMe-3800" "TMe-2033" "TMe-3766" "TMe-539"
#> [7] "TMe-2329" "TMe-3200" "TMe-2952" "TMe-171" "TMe-1732" "TMe-2568"
#> [13] "TMe-196" "TMe-40" "TMe-960"
#>
#> $III
#> [1] "TMe-3631" "TMe-425" "TMe-64" "TMe-155" "TMe-1804" "TMe-234" "TMe-3715"
#> [8] "TMe-2733" "TMe-1790"
#>
#> $IV
#> [1] "TMe-1988" "TMe-241" "TMe-2567" "TMe-1434" "TMe-812" "TMe-3581"
#> [7] "TMe-2924" "TMe-761" "TMe-3273" "TMe-3758" "TMe-2788" "TMe-2928"
#> [13] "TMe-2956" "TMe-1016" "TMe-280" "TMe-2059" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2853" "TMe-419" "TMe-2003" "TMe-712" "TMe-2355" "TMe-603"
#> [7] "TMe-2016" "TMe-730" "TMe-2750" "TMe-1265" "TMe-707" "TMe-2441"
#> [13] "TMe-2124" "TMe-755" "TMe-2753" "TMe-892" "TMe-2907" "TMe-588"
#> [19] "TMe-997" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1566" "TMe-2983" "TMe-1661" "TMe-1816" "TMe-1124" "TMe-1608" "TMe-2543"
#> [8] "TMe-751"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_eccentricity_out,
use.names = FALSE)) +
labs(title = "eccentricity")
# Peripheral Sampling by Maximal Eccentricity
sel_eccentricity_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "eccentricity")
sel_eccentricity_out
#> $I
#> [1] "TMe-2943" "TMe-3437" "TMe-3719" "TMe-3685" "TMe-3110" "TMe-1532"
#> [7] "TMe-3353" "TMe-1218" "TMe-306" "TMe-2027" "TMe-1117" "TMe-2103"
#> [13] "TMe-2967" "TMe-2810" "TMe-865" "TMe-1830"
#>
#> $II
#> [1] "TMe-2352" "TMe-369" "TMe-3800" "TMe-2033" "TMe-3766" "TMe-539"
#> [7] "TMe-2329" "TMe-3200" "TMe-2952" "TMe-171" "TMe-1732" "TMe-2568"
#> [13] "TMe-196" "TMe-40" "TMe-960"
#>
#> $III
#> [1] "TMe-3631" "TMe-425" "TMe-64" "TMe-155" "TMe-1804" "TMe-234" "TMe-3715"
#> [8] "TMe-2733" "TMe-1790"
#>
#> $IV
#> [1] "TMe-1988" "TMe-241" "TMe-2567" "TMe-1434" "TMe-812" "TMe-3581"
#> [7] "TMe-2924" "TMe-761" "TMe-3273" "TMe-3758" "TMe-2788" "TMe-2928"
#> [13] "TMe-2956" "TMe-1016" "TMe-280" "TMe-2059" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2853" "TMe-419" "TMe-2003" "TMe-712" "TMe-2355" "TMe-603"
#> [7] "TMe-2016" "TMe-730" "TMe-2750" "TMe-1265" "TMe-707" "TMe-2441"
#> [13] "TMe-2124" "TMe-755" "TMe-2753" "TMe-892" "TMe-2907" "TMe-588"
#> [19] "TMe-997" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1566" "TMe-2983" "TMe-1661" "TMe-1816" "TMe-1124" "TMe-1608" "TMe-2543"
#> [8] "TMe-751"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_eccentricity_out,
use.names = FALSE)) +
labs(title = "eccentricity")
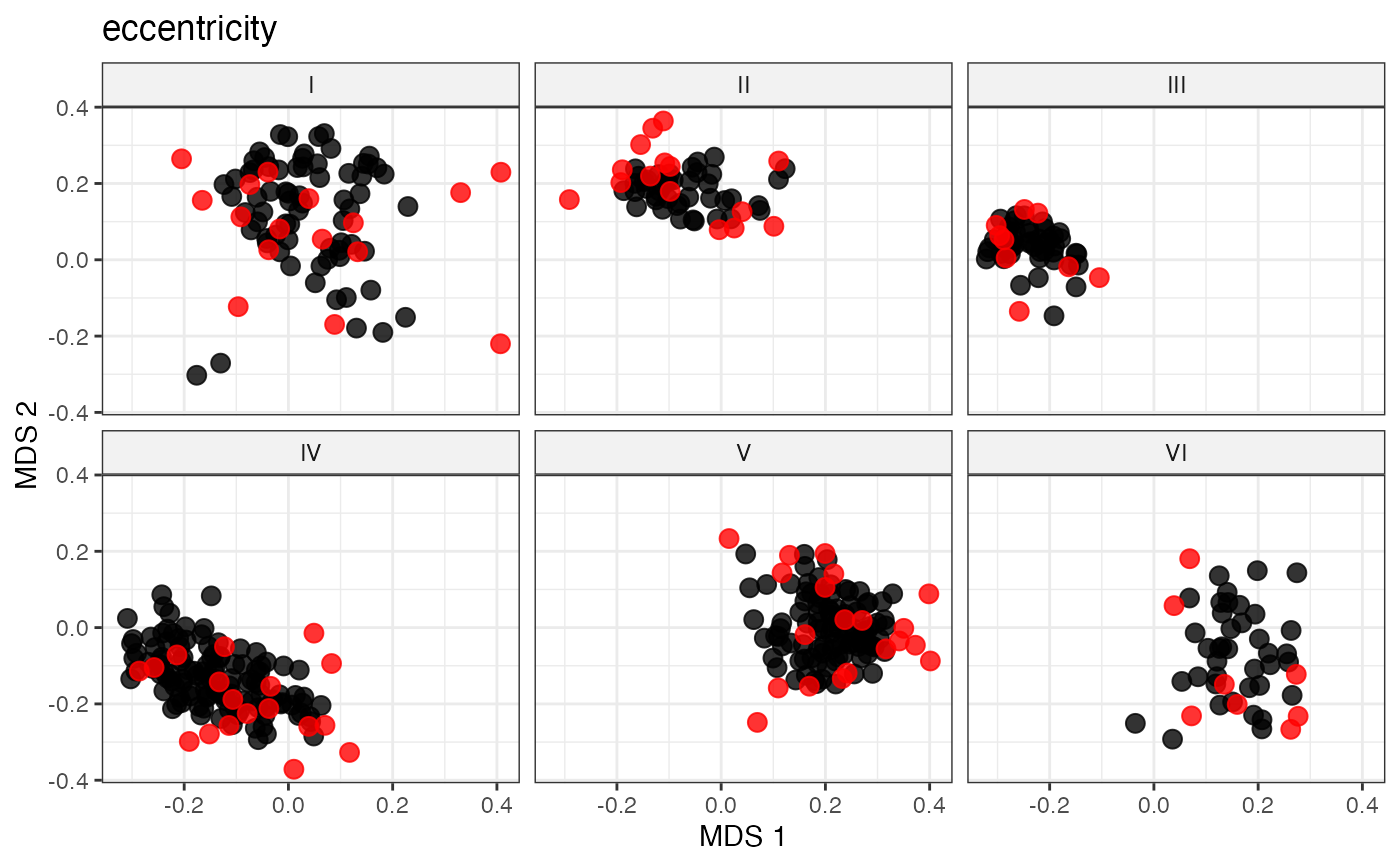 # Peripheral Sampling by Maximal Farness Centrality
sel_far_cent_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "farness.centrality")
sel_far_cent_out
#> $I
#> [1] "TMe-2967" "TMe-3685" "TMe-1425" "TMe-2943" "TMe-41" "TMe-3437"
#> [7] "TMe-3115" "TMe-3398" "TMe-500" "TMe-1190" "TMe-815" "TMe-569"
#> [13] "TMe-3726" "TMe-2027" "TMe-2934" "TMe-1830"
#>
#> $II
#> [1] "TMe-3766" "TMe-2952" "TMe-2033" "TMe-369" "TMe-3800" "TMe-3200"
#> [7] "TMe-674" "TMe-2329" "TMe-1385" "TMe-171" "TMe-2352" "TMe-2568"
#> [13] "TMe-3805" "TMe-1754" "TMe-3101"
#>
#> $III
#> [1] "TMe-3631" "TMe-234" "TMe-261" "TMe-425" "TMe-70" "TMe-381" "TMe-1804"
#> [8] "TMe-2374" "TMe-1790"
#>
#> $IV
#> [1] "TMe-241" "TMe-761" "TMe-812" "TMe-1434" "TMe-2924" "TMe-698"
#> [7] "TMe-1988" "TMe-1020" "TMe-3273" "TMe-2567" "TMe-2956" "TMe-427"
#> [13] "TMe-27" "TMe-3255" "TMe-1376" "TMe-2971" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-419" "TMe-2853" "TMe-2355" "TMe-723" "TMe-712" "TMe-603"
#> [7] "TMe-2907" "TMe-2003" "TMe-344" "TMe-532" "TMe-3329" "TMe-730"
#> [13] "TMe-1159" "TMe-2290" "TMe-2750" "TMe-98" "TMe-1375" "TMe-755"
#> [19] "TMe-1534" "TMe-2018"
#>
#> $VI
#> [1] "TMe-2983" "TMe-1403" "TMe-1124" "TMe-693" "TMe-751" "TMe-1992" "TMe-2791"
#> [8] "TMe-1035"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_far_cent_out,
use.names = FALSE)) +
labs(title = "farness.centrality")
# Peripheral Sampling by Maximal Farness Centrality
sel_far_cent_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "farness.centrality")
sel_far_cent_out
#> $I
#> [1] "TMe-2967" "TMe-3685" "TMe-1425" "TMe-2943" "TMe-41" "TMe-3437"
#> [7] "TMe-3115" "TMe-3398" "TMe-500" "TMe-1190" "TMe-815" "TMe-569"
#> [13] "TMe-3726" "TMe-2027" "TMe-2934" "TMe-1830"
#>
#> $II
#> [1] "TMe-3766" "TMe-2952" "TMe-2033" "TMe-369" "TMe-3800" "TMe-3200"
#> [7] "TMe-674" "TMe-2329" "TMe-1385" "TMe-171" "TMe-2352" "TMe-2568"
#> [13] "TMe-3805" "TMe-1754" "TMe-3101"
#>
#> $III
#> [1] "TMe-3631" "TMe-234" "TMe-261" "TMe-425" "TMe-70" "TMe-381" "TMe-1804"
#> [8] "TMe-2374" "TMe-1790"
#>
#> $IV
#> [1] "TMe-241" "TMe-761" "TMe-812" "TMe-1434" "TMe-2924" "TMe-698"
#> [7] "TMe-1988" "TMe-1020" "TMe-3273" "TMe-2567" "TMe-2956" "TMe-427"
#> [13] "TMe-27" "TMe-3255" "TMe-1376" "TMe-2971" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-419" "TMe-2853" "TMe-2355" "TMe-723" "TMe-712" "TMe-603"
#> [7] "TMe-2907" "TMe-2003" "TMe-344" "TMe-532" "TMe-3329" "TMe-730"
#> [13] "TMe-1159" "TMe-2290" "TMe-2750" "TMe-98" "TMe-1375" "TMe-755"
#> [19] "TMe-1534" "TMe-2018"
#>
#> $VI
#> [1] "TMe-2983" "TMe-1403" "TMe-1124" "TMe-693" "TMe-751" "TMe-1992" "TMe-2791"
#> [8] "TMe-1035"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_far_cent_out,
use.names = FALSE)) +
labs(title = "farness.centrality")
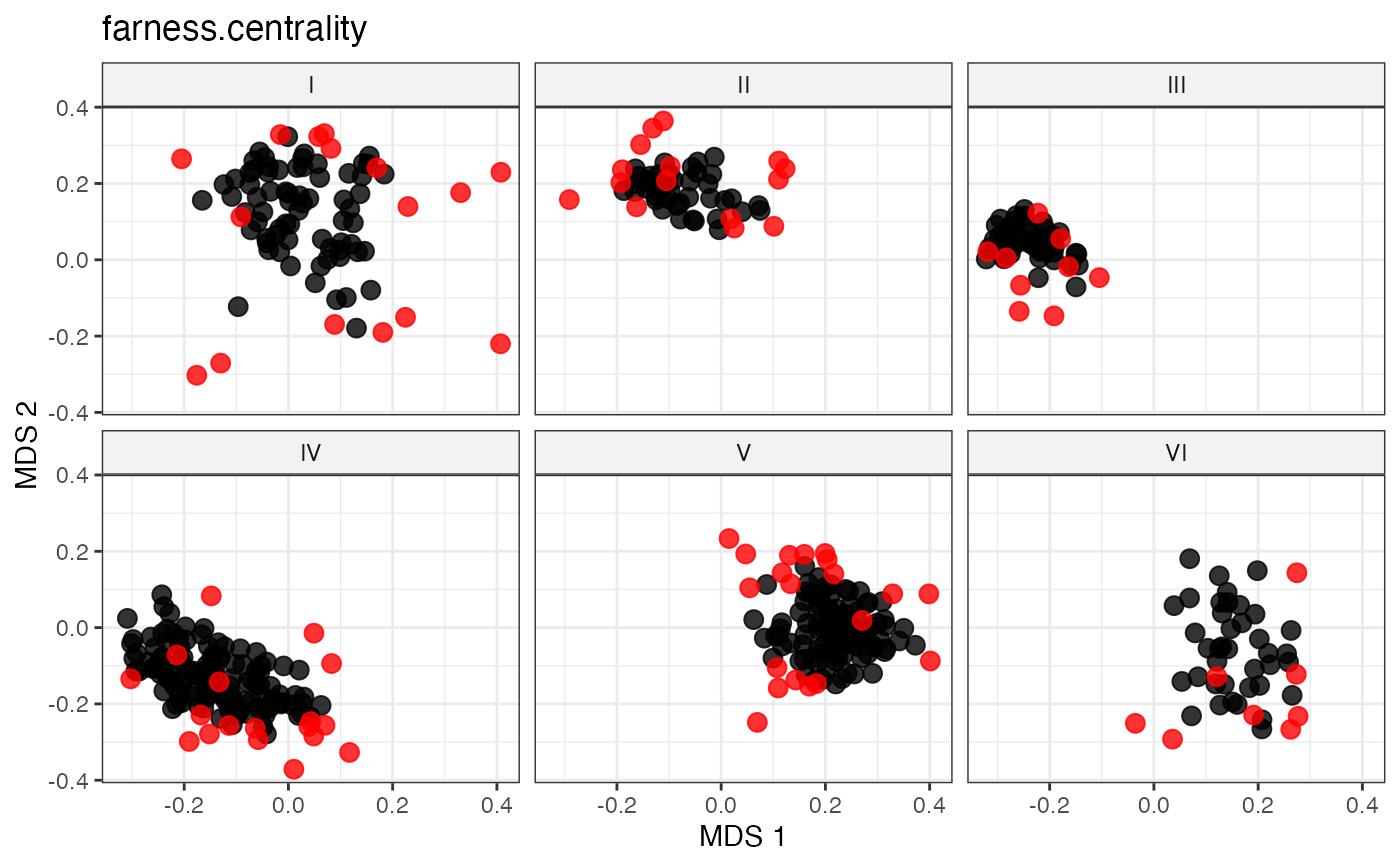 #~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Fetch selected accessions by space-Filling/coverage methods
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Space-Filling Sampling via the Kennard-Stone Algorithm
sel_ks_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "kennard.stone")
sel_ks_out
#> $I
#> [1] "TMe-3111" "TMe-3130" "TMe-44" "TMe-3623" "TMe-1930" "TMe-1451"
#> [7] "TMe-2934" "TMe-3481" "TMe-3051" "TMe-1581" "TMe-2237" "TMe-3548"
#> [13] "TMe-1589" "TMe-1096" "TMe-3719" "TMe-1830"
#>
#> $II
#> [1] "TMe-1732" "TMe-2715" "TMe-681" "TMe-2211" "TMe-796" "TMe-3366"
#> [7] "TMe-196" "TMe-1754" "TMe-2021" "TMe-3093" "TMe-455" "TMe-3800"
#> [13] "TMe-2995" "TMe-2997" "TMe-3495"
#>
#> $III
#> [1] "TMe-2374" "TMe-830" "TMe-148" "TMe-2086" "TMe-425" "TMe-1939" "TMe-3207"
#> [8] "TMe-2161" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2039" "TMe-57" "TMe-2567" "TMe-3538" "TMe-182" "TMe-1456"
#> [7] "TMe-3757" "TMe-1996" "TMe-2807" "TMe-3409" "TMe-3581" "TMe-3459"
#> [13] "TMe-3144" "TMe-2839" "TMe-78" "TMe-737" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2750" "TMe-688" "TMe-247" "TMe-2016" "TMe-423" "TMe-419"
#> [7] "TMe-312" "TMe-256" "TMe-723" "TMe-288" "TMe-920" "TMe-348"
#> [13] "TMe-2151" "TMe-1012" "TMe-682" "TMe-487" "TMe-1268" "TMe-2290"
#> [19] "TMe-2441" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1413" "TMe-1124" "TMe-2983" "TMe-936" "TMe-1403" "TMe-1035" "TMe-968"
#> [8] "TMe-2818"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_ks_out,
use.names = FALSE)) +
labs(title = "kennard.stone")
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Fetch selected accessions by space-Filling/coverage methods
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Space-Filling Sampling via the Kennard-Stone Algorithm
sel_ks_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "kennard.stone")
sel_ks_out
#> $I
#> [1] "TMe-3111" "TMe-3130" "TMe-44" "TMe-3623" "TMe-1930" "TMe-1451"
#> [7] "TMe-2934" "TMe-3481" "TMe-3051" "TMe-1581" "TMe-2237" "TMe-3548"
#> [13] "TMe-1589" "TMe-1096" "TMe-3719" "TMe-1830"
#>
#> $II
#> [1] "TMe-1732" "TMe-2715" "TMe-681" "TMe-2211" "TMe-796" "TMe-3366"
#> [7] "TMe-196" "TMe-1754" "TMe-2021" "TMe-3093" "TMe-455" "TMe-3800"
#> [13] "TMe-2995" "TMe-2997" "TMe-3495"
#>
#> $III
#> [1] "TMe-2374" "TMe-830" "TMe-148" "TMe-2086" "TMe-425" "TMe-1939" "TMe-3207"
#> [8] "TMe-2161" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2039" "TMe-57" "TMe-2567" "TMe-3538" "TMe-182" "TMe-1456"
#> [7] "TMe-3757" "TMe-1996" "TMe-2807" "TMe-3409" "TMe-3581" "TMe-3459"
#> [13] "TMe-3144" "TMe-2839" "TMe-78" "TMe-737" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2750" "TMe-688" "TMe-247" "TMe-2016" "TMe-423" "TMe-419"
#> [7] "TMe-312" "TMe-256" "TMe-723" "TMe-288" "TMe-920" "TMe-348"
#> [13] "TMe-2151" "TMe-1012" "TMe-682" "TMe-487" "TMe-1268" "TMe-2290"
#> [19] "TMe-2441" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1413" "TMe-1124" "TMe-2983" "TMe-936" "TMe-1403" "TMe-1035" "TMe-968"
#> [8] "TMe-2818"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_ks_out,
use.names = FALSE)) +
labs(title = "kennard.stone")
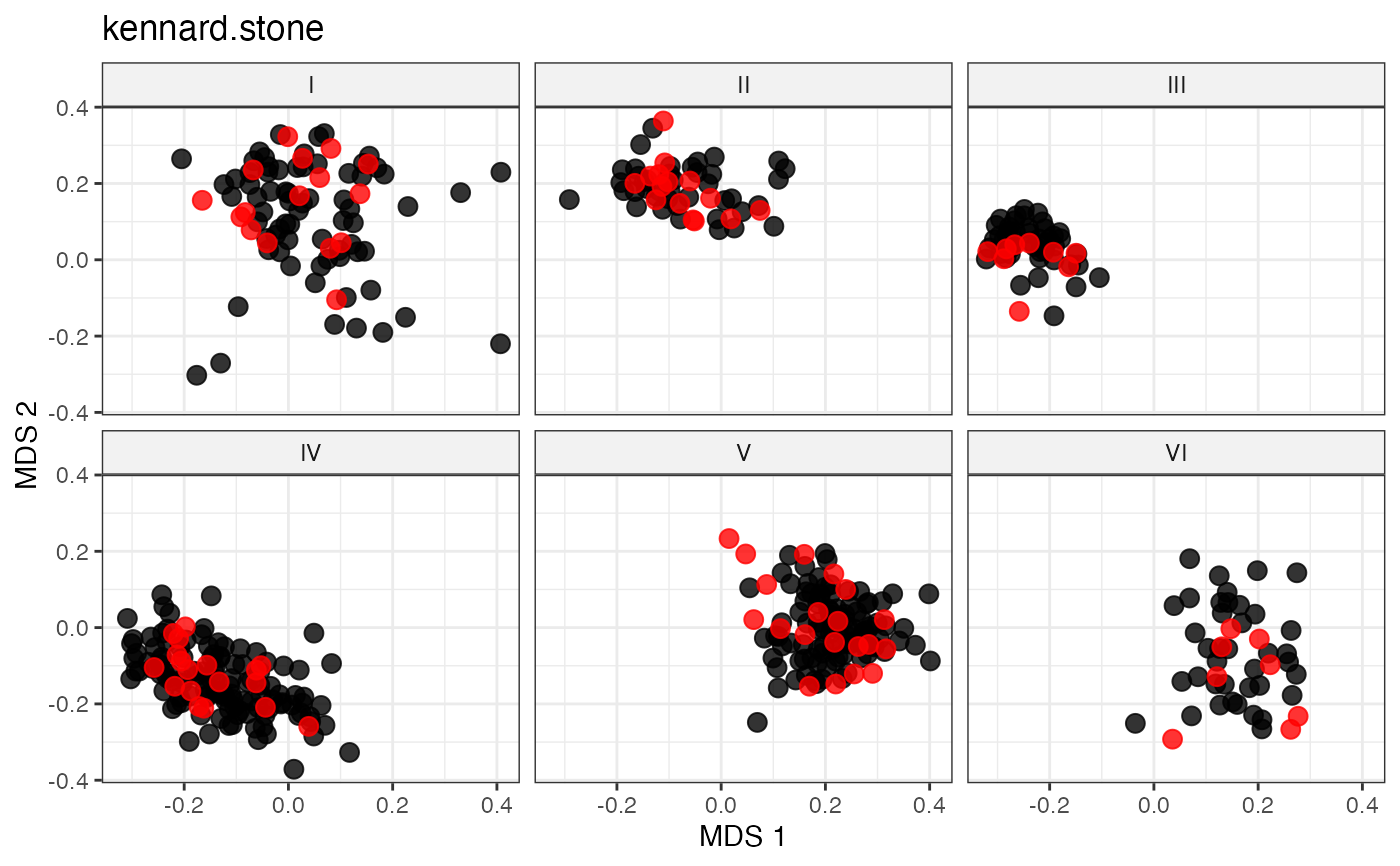 # Space-Filling Sampling via the DUPLEX Algorithm
sel_duplex_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "duplex")
sel_duplex_out
#> $I
#> [1] "TMe-3111" "TMe-3130" "TMe-44" "TMe-3623" "TMe-1930" "TMe-1451"
#> [7] "TMe-2934" "TMe-3481" "TMe-3051" "TMe-1581" "TMe-2237" "TMe-3548"
#> [13] "TMe-1589" "TMe-1096" "TMe-3719" "TMe-1830"
#>
#> $II
#> [1] "TMe-1732" "TMe-2715" "TMe-681" "TMe-2211" "TMe-796" "TMe-3366"
#> [7] "TMe-1754" "TMe-2021" "TMe-3093" "TMe-455" "TMe-2995" "TMe-674"
#> [13] "TMe-2997" "TMe-3495" "TMe-2952"
#>
#> $III
#> [1] "TMe-2374" "TMe-830" "TMe-148" "TMe-2086" "TMe-425" "TMe-1939" "TMe-2161"
#> [8] "TMe-234" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2039" "TMe-57" "TMe-2567" "TMe-3538" "TMe-182" "TMe-1456"
#> [7] "TMe-3757" "TMe-1996" "TMe-2807" "TMe-3581" "TMe-3459" "TMe-2839"
#> [13] "TMe-78" "TMe-737" "TMe-25" "TMe-3542" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2750" "TMe-688" "TMe-247" "TMe-2016" "TMe-423" "TMe-419"
#> [7] "TMe-312" "TMe-723" "TMe-920" "TMe-348" "TMe-2151" "TMe-1012"
#> [13] "TMe-682" "TMe-487" "TMe-2290" "TMe-362" "TMe-1037" "TMe-1401"
#> [19] "TMe-600" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1413" "TMe-1124" "TMe-2983" "TMe-1302" "TMe-1608" "TMe-505" "TMe-1676"
#> [8] "TMe-1816"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_duplex_out,
use.names = FALSE)) +
labs(title = "duplex")
# Space-Filling Sampling via the DUPLEX Algorithm
sel_duplex_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "duplex")
sel_duplex_out
#> $I
#> [1] "TMe-3111" "TMe-3130" "TMe-44" "TMe-3623" "TMe-1930" "TMe-1451"
#> [7] "TMe-2934" "TMe-3481" "TMe-3051" "TMe-1581" "TMe-2237" "TMe-3548"
#> [13] "TMe-1589" "TMe-1096" "TMe-3719" "TMe-1830"
#>
#> $II
#> [1] "TMe-1732" "TMe-2715" "TMe-681" "TMe-2211" "TMe-796" "TMe-3366"
#> [7] "TMe-1754" "TMe-2021" "TMe-3093" "TMe-455" "TMe-2995" "TMe-674"
#> [13] "TMe-2997" "TMe-3495" "TMe-2952"
#>
#> $III
#> [1] "TMe-2374" "TMe-830" "TMe-148" "TMe-2086" "TMe-425" "TMe-1939" "TMe-2161"
#> [8] "TMe-234" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2039" "TMe-57" "TMe-2567" "TMe-3538" "TMe-182" "TMe-1456"
#> [7] "TMe-3757" "TMe-1996" "TMe-2807" "TMe-3581" "TMe-3459" "TMe-2839"
#> [13] "TMe-78" "TMe-737" "TMe-25" "TMe-3542" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2750" "TMe-688" "TMe-247" "TMe-2016" "TMe-423" "TMe-419"
#> [7] "TMe-312" "TMe-723" "TMe-920" "TMe-348" "TMe-2151" "TMe-1012"
#> [13] "TMe-682" "TMe-487" "TMe-2290" "TMe-362" "TMe-1037" "TMe-1401"
#> [19] "TMe-600" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1413" "TMe-1124" "TMe-2983" "TMe-1302" "TMe-1608" "TMe-505" "TMe-1676"
#> [8] "TMe-1816"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_duplex_out,
use.names = FALSE)) +
labs(title = "duplex")
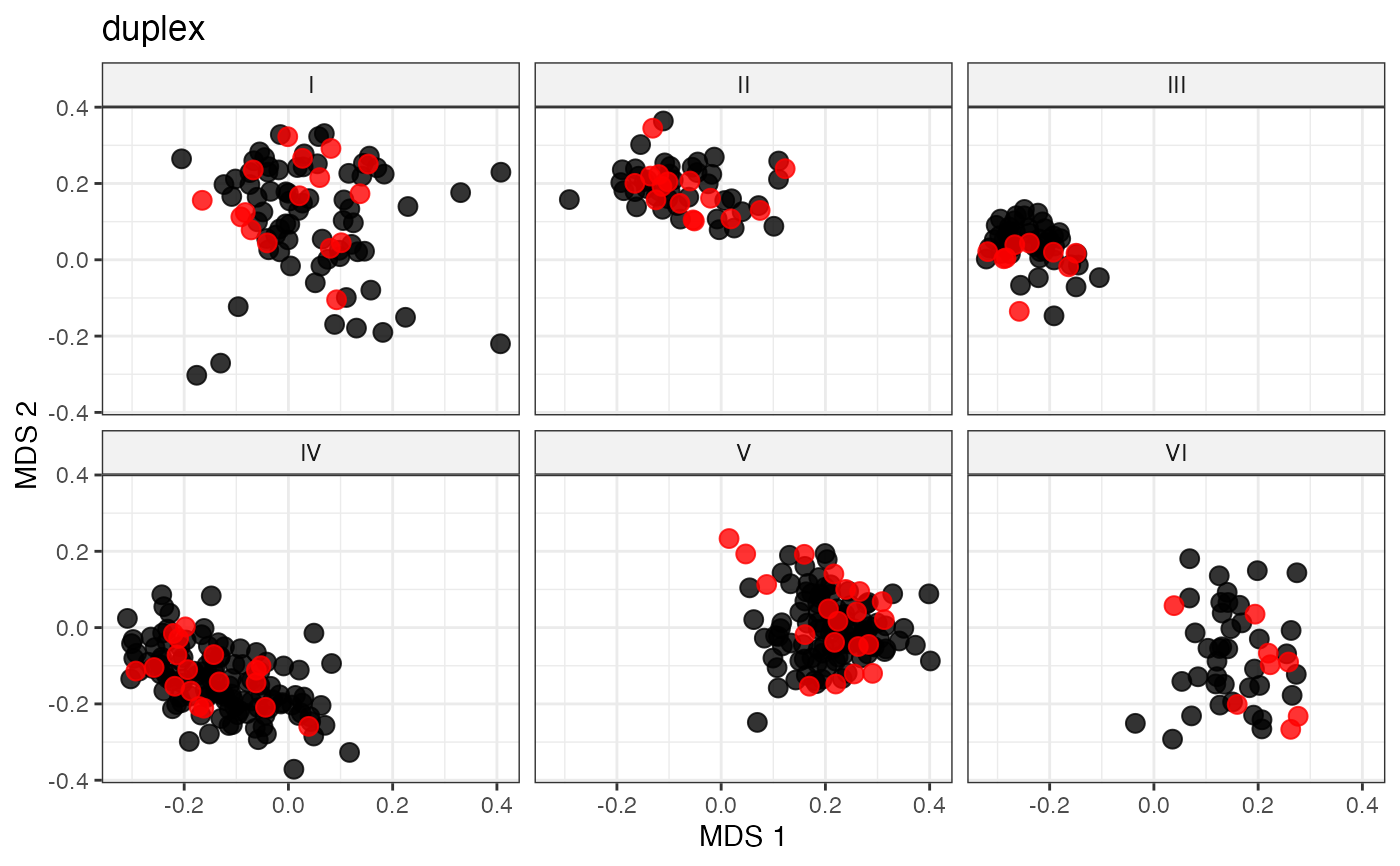 # Space-Filling Sampling via the Honigs Algorithm
sel_honigs_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "honigs")
sel_honigs_out
#> $I
#> [1] "TMe-3437" "TMe-2943" "TMe-1425" "TMe-2967" "TMe-815" "TMe-3685"
#> [7] "TMe-3353" "TMe-41" "TMe-1190" "TMe-2975" "TMe-2027" "TMe-2785"
#> [13] "TMe-2934" "TMe-1922" "TMe-1170" "TMe-1830"
#>
#> $II
#> [1] "TMe-369" "TMe-2352" "TMe-2952" "TMe-171" "TMe-3101" "TMe-2568"
#> [7] "TMe-2033" "TMe-3200" "TMe-3547" "TMe-251" "TMe-3800" "TMe-74"
#> [13] "TMe-3766" "TMe-2211" "TMe-1385"
#>
#> $III
#> [1] "TMe-425" "TMe-3631" "TMe-234" "TMe-1819" "TMe-1176" "TMe-3715" "TMe-3569"
#> [8] "TMe-70" "TMe-1790"
#>
#> $IV
#> [1] "TMe-241" "TMe-1988" "TMe-25" "TMe-812" "TMe-1434" "TMe-427"
#> [7] "TMe-2958" "TMe-761" "TMe-460" "TMe-2567" "TMe-737" "TMe-619"
#> [13] "TMe-3189" "TMe-280" "TMe-368" "TMe-3527" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-419" "TMe-2853" "TMe-224" "TMe-487" "TMe-603" "TMe-723"
#> [7] "TMe-2855" "TMe-3329" "TMe-1220" "TMe-2290" "TMe-1934" "TMe-755"
#> [13] "TMe-362" "TMe-1375" "TMe-712" "TMe-256" "TMe-2355" "TMe-816"
#> [19] "TMe-1401" "TMe-2018"
#>
#> $VI
#> [1] "TMe-2983" "TMe-1566" "TMe-310" "TMe-1124" "TMe-1816" "TMe-2791" "TMe-1403"
#> [8] "TMe-751"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_honigs_out,
use.names = FALSE)) +
labs(title = "honigs")
# Space-Filling Sampling via the Honigs Algorithm
sel_honigs_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "honigs")
sel_honigs_out
#> $I
#> [1] "TMe-3437" "TMe-2943" "TMe-1425" "TMe-2967" "TMe-815" "TMe-3685"
#> [7] "TMe-3353" "TMe-41" "TMe-1190" "TMe-2975" "TMe-2027" "TMe-2785"
#> [13] "TMe-2934" "TMe-1922" "TMe-1170" "TMe-1830"
#>
#> $II
#> [1] "TMe-369" "TMe-2352" "TMe-2952" "TMe-171" "TMe-3101" "TMe-2568"
#> [7] "TMe-2033" "TMe-3200" "TMe-3547" "TMe-251" "TMe-3800" "TMe-74"
#> [13] "TMe-3766" "TMe-2211" "TMe-1385"
#>
#> $III
#> [1] "TMe-425" "TMe-3631" "TMe-234" "TMe-1819" "TMe-1176" "TMe-3715" "TMe-3569"
#> [8] "TMe-70" "TMe-1790"
#>
#> $IV
#> [1] "TMe-241" "TMe-1988" "TMe-25" "TMe-812" "TMe-1434" "TMe-427"
#> [7] "TMe-2958" "TMe-761" "TMe-460" "TMe-2567" "TMe-737" "TMe-619"
#> [13] "TMe-3189" "TMe-280" "TMe-368" "TMe-3527" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-419" "TMe-2853" "TMe-224" "TMe-487" "TMe-603" "TMe-723"
#> [7] "TMe-2855" "TMe-3329" "TMe-1220" "TMe-2290" "TMe-1934" "TMe-755"
#> [13] "TMe-362" "TMe-1375" "TMe-712" "TMe-256" "TMe-2355" "TMe-816"
#> [19] "TMe-1401" "TMe-2018"
#>
#> $VI
#> [1] "TMe-2983" "TMe-1566" "TMe-310" "TMe-1124" "TMe-1816" "TMe-2791" "TMe-1403"
#> [8] "TMe-751"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_honigs_out,
use.names = FALSE)) +
labs(title = "honigs")
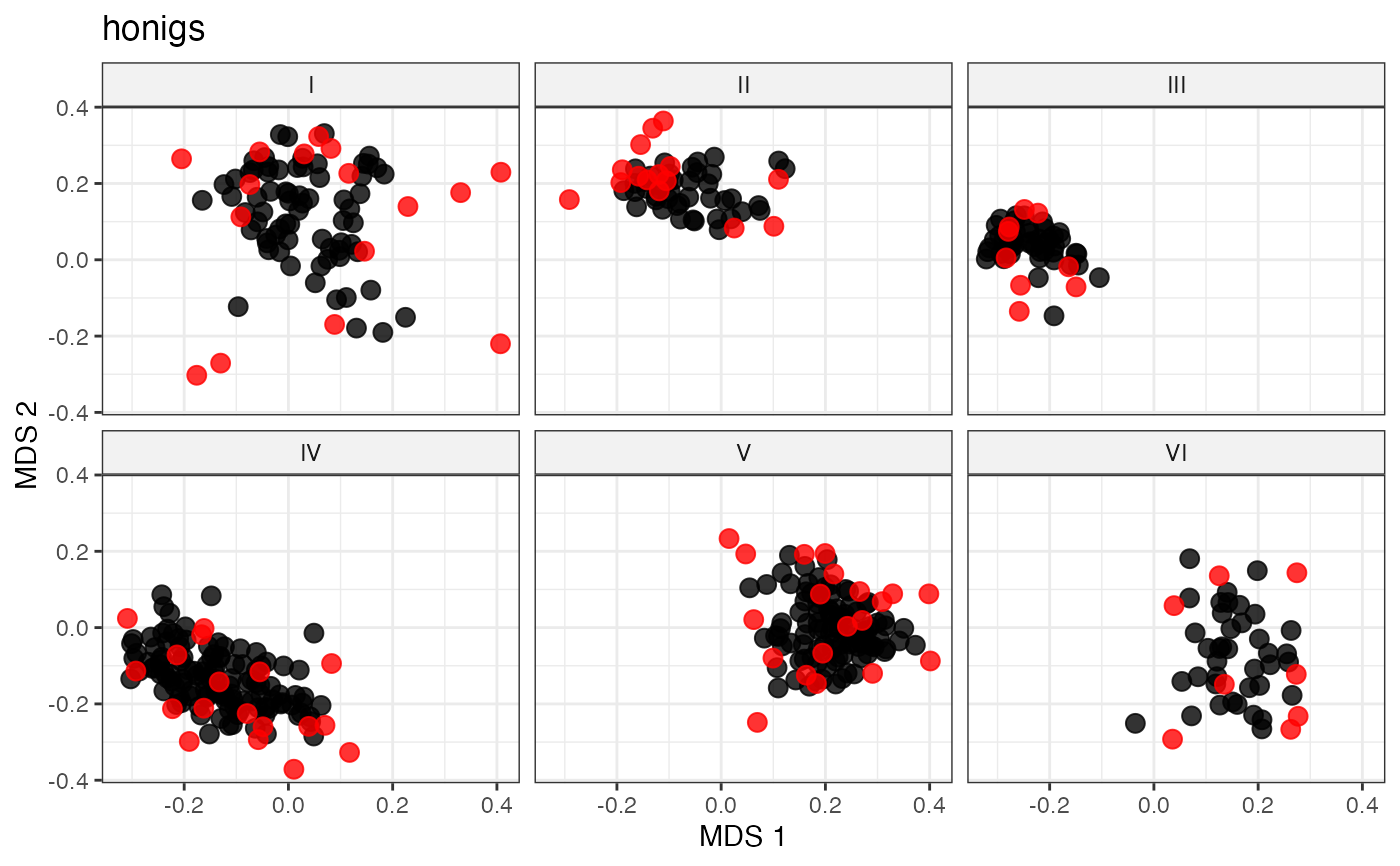 # Space-Filling Sampling via Farthest-Point (Max-Min) Algorithm
sel_far_pt_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "farthest.point")
sel_far_pt_out
#> $I
#> [1] "TMe-882" "TMe-2967" "TMe-3685" "TMe-41" "TMe-2785" "TMe-1922"
#> [7] "TMe-778" "TMe-2943" "TMe-1425" "TMe-3252" "TMe-1589" "TMe-1190"
#> [13] "TMe-3112" "TMe-3437" "TMe-3353" "TMe-1830"
#>
#> $II
#> [1] "TMe-1698" "TMe-369" "TMe-2568" "TMe-2033" "TMe-3800" "TMe-3200"
#> [7] "TMe-2329" "TMe-251" "TMe-1385" "TMe-3101" "TMe-2952" "TMe-2352"
#> [13] "TMe-960" "TMe-3284" "TMe-3258"
#>
#> $III
#> [1] "TMe-14" "TMe-261" "TMe-381" "TMe-234" "TMe-3007" "TMe-1863" "TMe-1819"
#> [8] "TMe-1738" "TMe-1790"
#>
#> $IV
#> [1] "TMe-108" "TMe-241" "TMe-2240" "TMe-1434" "TMe-3269" "TMe-698"
#> [7] "TMe-27" "TMe-1148" "TMe-78" "TMe-1020" "TMe-3255" "TMe-372"
#> [13] "TMe-3196" "TMe-3428" "TMe-812" "TMe-1988" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1273" "TMe-419" "TMe-1159" "TMe-712" "TMe-256" "TMe-920"
#> [7] "TMe-723" "TMe-2003" "TMe-3329" "TMe-487" "TMe-532" "TMe-2355"
#> [13] "TMe-877" "TMe-1534" "TMe-1934" "TMe-2853" "TMe-603" "TMe-439"
#> [19] "TMe-730" "TMe-2018"
#>
#> $VI
#> [1] "TMe-3177" "TMe-1124" "TMe-2983" "TMe-2543" "TMe-693" "TMe-751" "TMe-2791"
#> [8] "TMe-1816"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_far_pt_out,
use.names = FALSE)) +
labs(title = "farthest.point")
# Space-Filling Sampling via Farthest-Point (Max-Min) Algorithm
sel_far_pt_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "farthest.point")
sel_far_pt_out
#> $I
#> [1] "TMe-882" "TMe-2967" "TMe-3685" "TMe-41" "TMe-2785" "TMe-1922"
#> [7] "TMe-778" "TMe-2943" "TMe-1425" "TMe-3252" "TMe-1589" "TMe-1190"
#> [13] "TMe-3112" "TMe-3437" "TMe-3353" "TMe-1830"
#>
#> $II
#> [1] "TMe-1698" "TMe-369" "TMe-2568" "TMe-2033" "TMe-3800" "TMe-3200"
#> [7] "TMe-2329" "TMe-251" "TMe-1385" "TMe-3101" "TMe-2952" "TMe-2352"
#> [13] "TMe-960" "TMe-3284" "TMe-3258"
#>
#> $III
#> [1] "TMe-14" "TMe-261" "TMe-381" "TMe-234" "TMe-3007" "TMe-1863" "TMe-1819"
#> [8] "TMe-1738" "TMe-1790"
#>
#> $IV
#> [1] "TMe-108" "TMe-241" "TMe-2240" "TMe-1434" "TMe-3269" "TMe-698"
#> [7] "TMe-27" "TMe-1148" "TMe-78" "TMe-1020" "TMe-3255" "TMe-372"
#> [13] "TMe-3196" "TMe-3428" "TMe-812" "TMe-1988" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1273" "TMe-419" "TMe-1159" "TMe-712" "TMe-256" "TMe-920"
#> [7] "TMe-723" "TMe-2003" "TMe-3329" "TMe-487" "TMe-532" "TMe-2355"
#> [13] "TMe-877" "TMe-1534" "TMe-1934" "TMe-2853" "TMe-603" "TMe-439"
#> [19] "TMe-730" "TMe-2018"
#>
#> $VI
#> [1] "TMe-3177" "TMe-1124" "TMe-2983" "TMe-2543" "TMe-693" "TMe-751" "TMe-2791"
#> [8] "TMe-1816"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_far_pt_out,
use.names = FALSE)) +
labs(title = "farthest.point")
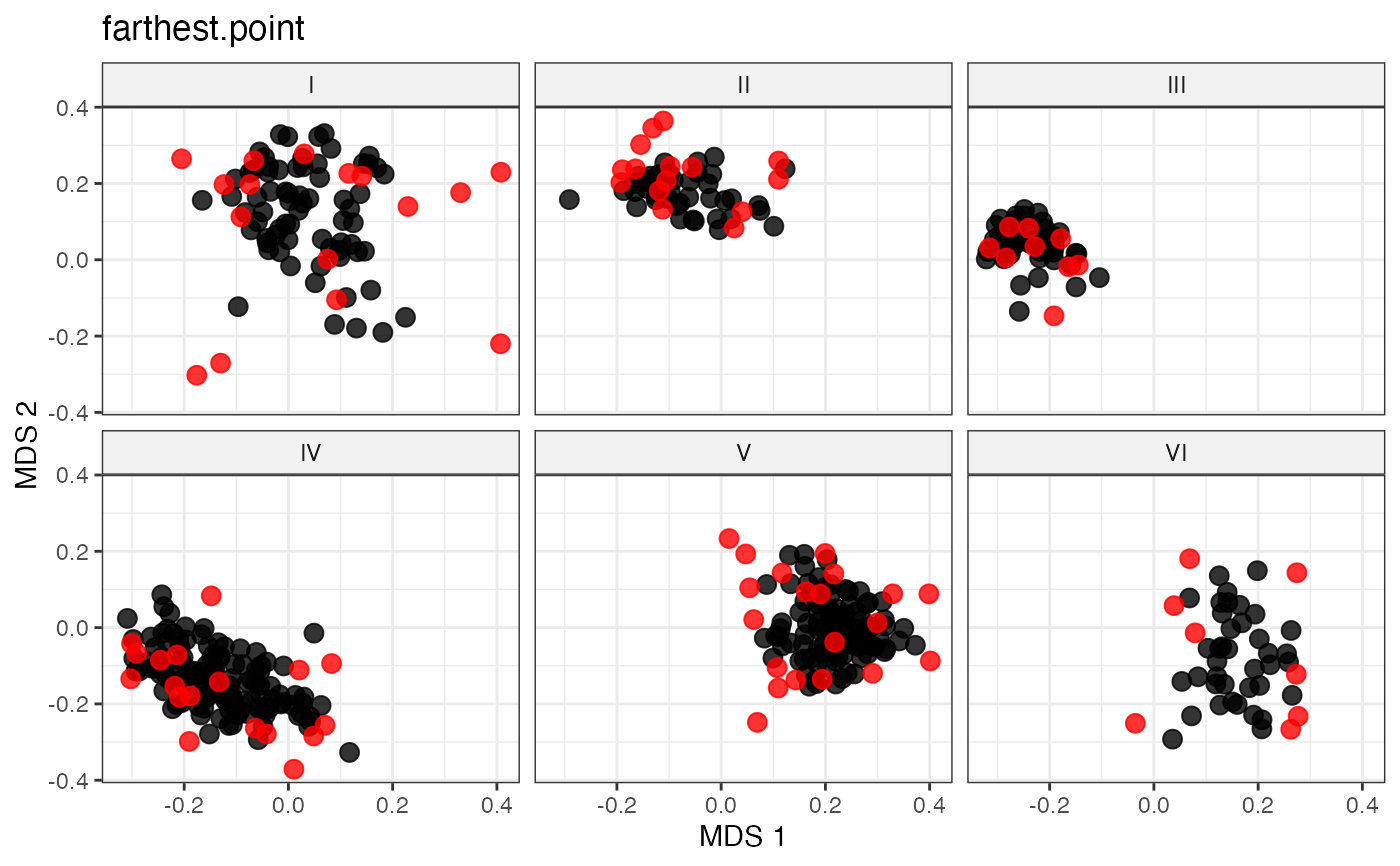 #~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Fetch selected accessions by density based methods
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Density-Based Sampling by Minimal Nearest-Neighbour Distance
sel_nn_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "nearest.neighbour")
sel_nn_out
#> $I
#> [1] "TMe-2103" "TMe-2152" "TMe-1717" "TMe-1117" "TMe-910" "TMe-2066"
#> [7] "TMe-865" "TMe-1091" "TMe-1823" "TMe-937" "TMe-1973" "TMe-2944"
#> [13] "TMe-3553" "TMe-3419" "TMe-2964" "TMe-1830"
#>
#> $II
#> [1] "TMe-890" "TMe-455" "TMe-2412" "TMe-2021" "TMe-2258" "TMe-2000"
#> [7] "TMe-2715" "TMe-681" "TMe-2757" "TMe-1474" "TMe-3495" "TMe-3239"
#> [13] "TMe-2951" "TMe-409" "TMe-3258"
#>
#> $III
#> [1] "TMe-3750" "TMe-3148" "TMe-3007" "TMe-3207" "TMe-1198" "TMe-2502" "TMe-1965"
#> [8] "TMe-2394" "TMe-1790"
#>
#> $IV
#> [1] "TMe-1139" "TMe-2247" "TMe-428" "TMe-1765" "TMe-3232" "TMe-2039"
#> [7] "TMe-1123" "TMe-108" "TMe-525" "TMe-1155" "TMe-318" "TMe-3459"
#> [13] "TMe-3068" "TMe-2375" "TMe-191" "TMe-3538" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2441" "TMe-1160" "TMe-2124" "TMe-1979" "TMe-360" "TMe-312"
#> [7] "TMe-348" "TMe-307" "TMe-745" "TMe-1299" "TMe-1188" "TMe-647"
#> [13] "TMe-332" "TMe-1680" "TMe-1234" "TMe-1414" "TMe-2271" "TMe-870"
#> [19] "TMe-645" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-505" "TMe-1608" "TMe-1744" "TMe-936" "TMe-1832" "TMe-968"
#> [8] "TMe-1062"
#>
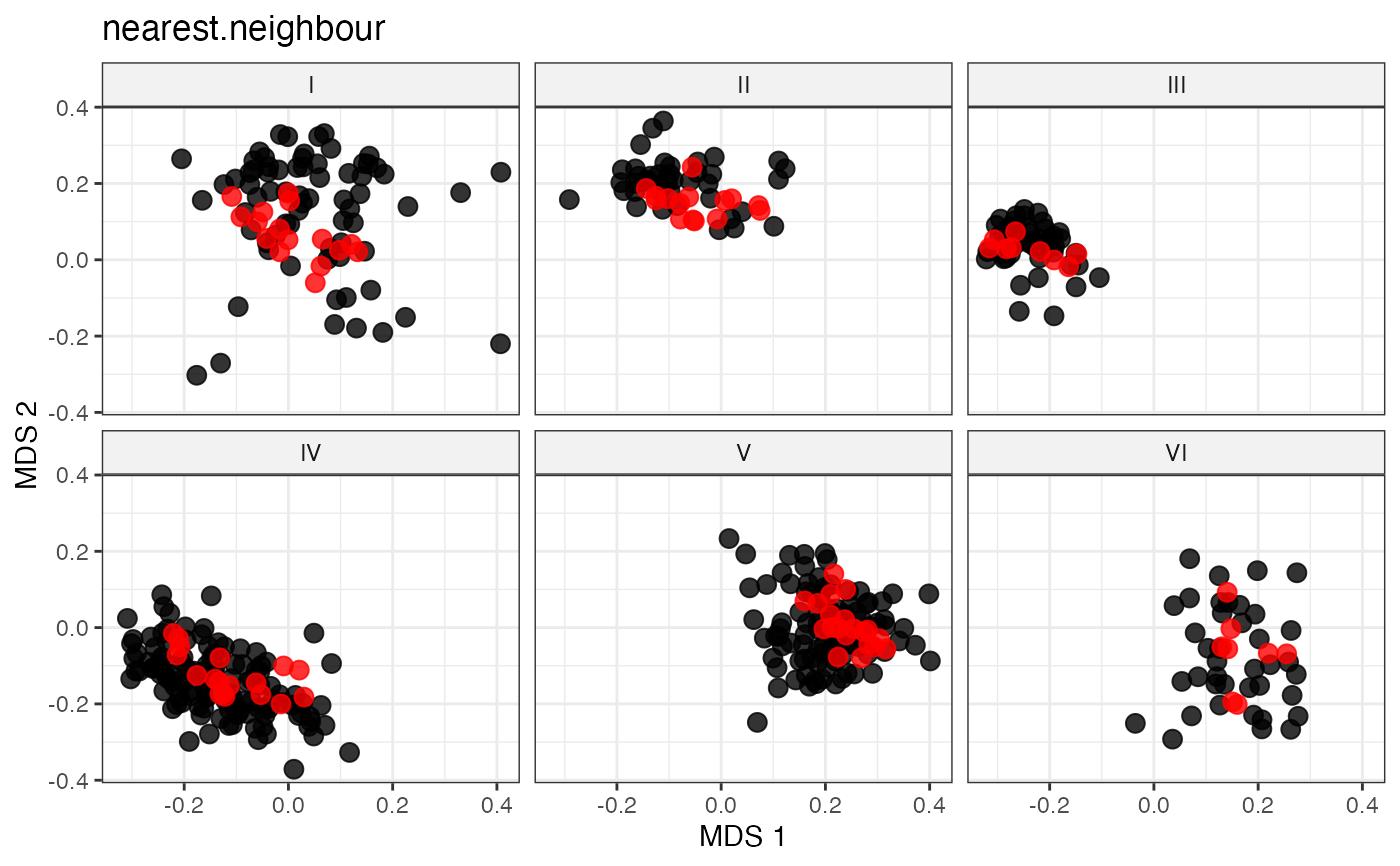
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_nn_out,
use.names = FALSE)) +
labs(title = "nearest.neighbour")
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Fetch selected accessions by density based methods
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Density-Based Sampling by Minimal Nearest-Neighbour Distance
sel_nn_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "nearest.neighbour")
sel_nn_out
#> $I
#> [1] "TMe-2103" "TMe-2152" "TMe-1717" "TMe-1117" "TMe-910" "TMe-2066"
#> [7] "TMe-865" "TMe-1091" "TMe-1823" "TMe-937" "TMe-1973" "TMe-2944"
#> [13] "TMe-3553" "TMe-3419" "TMe-2964" "TMe-1830"
#>
#> $II
#> [1] "TMe-890" "TMe-455" "TMe-2412" "TMe-2021" "TMe-2258" "TMe-2000"
#> [7] "TMe-2715" "TMe-681" "TMe-2757" "TMe-1474" "TMe-3495" "TMe-3239"
#> [13] "TMe-2951" "TMe-409" "TMe-3258"
#>
#> $III
#> [1] "TMe-3750" "TMe-3148" "TMe-3007" "TMe-3207" "TMe-1198" "TMe-2502" "TMe-1965"
#> [8] "TMe-2394" "TMe-1790"
#>
#> $IV
#> [1] "TMe-1139" "TMe-2247" "TMe-428" "TMe-1765" "TMe-3232" "TMe-2039"
#> [7] "TMe-1123" "TMe-108" "TMe-525" "TMe-1155" "TMe-318" "TMe-3459"
#> [13] "TMe-3068" "TMe-2375" "TMe-191" "TMe-3538" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2441" "TMe-1160" "TMe-2124" "TMe-1979" "TMe-360" "TMe-312"
#> [7] "TMe-348" "TMe-307" "TMe-745" "TMe-1299" "TMe-1188" "TMe-647"
#> [13] "TMe-332" "TMe-1680" "TMe-1234" "TMe-1414" "TMe-2271" "TMe-870"
#> [19] "TMe-645" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-505" "TMe-1608" "TMe-1744" "TMe-936" "TMe-1832" "TMe-968"
#> [8] "TMe-1062"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_nn_out,
use.names = FALSE)) +
labs(title = "nearest.neighbour")
 #~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Fetch selected accessions by cluster based methods
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Globally Optimal Medoid Sampling via Partitioning Around Medoids (PAM)
sel_pam_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "optim.medoid")
sel_pam_out
#> $I
#> [1] "TMe-2103" "TMe-28" "TMe-44" "TMe-3419" "TMe-1823" "TMe-952"
#> [7] "TMe-2027" "TMe-882" "TMe-910" "TMe-2944" "TMe-1091" "TMe-566"
#> [13] "TMe-3694" "TMe-3115" "TMe-1425" "TMe-1830"
#>
#> $II
#> [1] "TMe-2021" "TMe-681" "TMe-890" "TMe-2951" "TMe-3557" "TMe-3239"
#> [7] "TMe-1385" "TMe-2352" "TMe-1831" "TMe-1754" "TMe-1668" "TMe-409"
#> [13] "TMe-2033" "TMe-2611" "TMe-3200"
#>
#> $III
#> [1] "TMe-3007" "TMe-2394" "TMe-161" "TMe-2356" "TMe-123" "TMe-3335" "TMe-617"
#> [8] "TMe-2748" "TMe-1790"
#>
#> $IV
#> [1] "TMe-594" "TMe-12" "TMe-3378" "TMe-3409" "TMe-1027" "TMe-1155"
#> [7] "TMe-428" "TMe-170" "TMe-2947" "TMe-1139" "TMe-57" "TMe-2458"
#> [13] "TMe-525" "TMe-3068" "TMe-3265" "TMe-3562" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1979" "TMe-629" "TMe-344" "TMe-1234" "TMe-1788" "TMe-288"
#> [7] "TMe-1160" "TMe-954" "TMe-439" "TMe-245" "TMe-645" "TMe-2907"
#> [13] "TMe-647" "TMe-487" "TMe-723" "TMe-1622" "TMe-1880" "TMe-1559"
#> [19] "TMe-2853" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1608" "TMe-2196" "TMe-936" "TMe-1503" "TMe-1217" "TMe-620"
#> [8] "TMe-1445"
#>
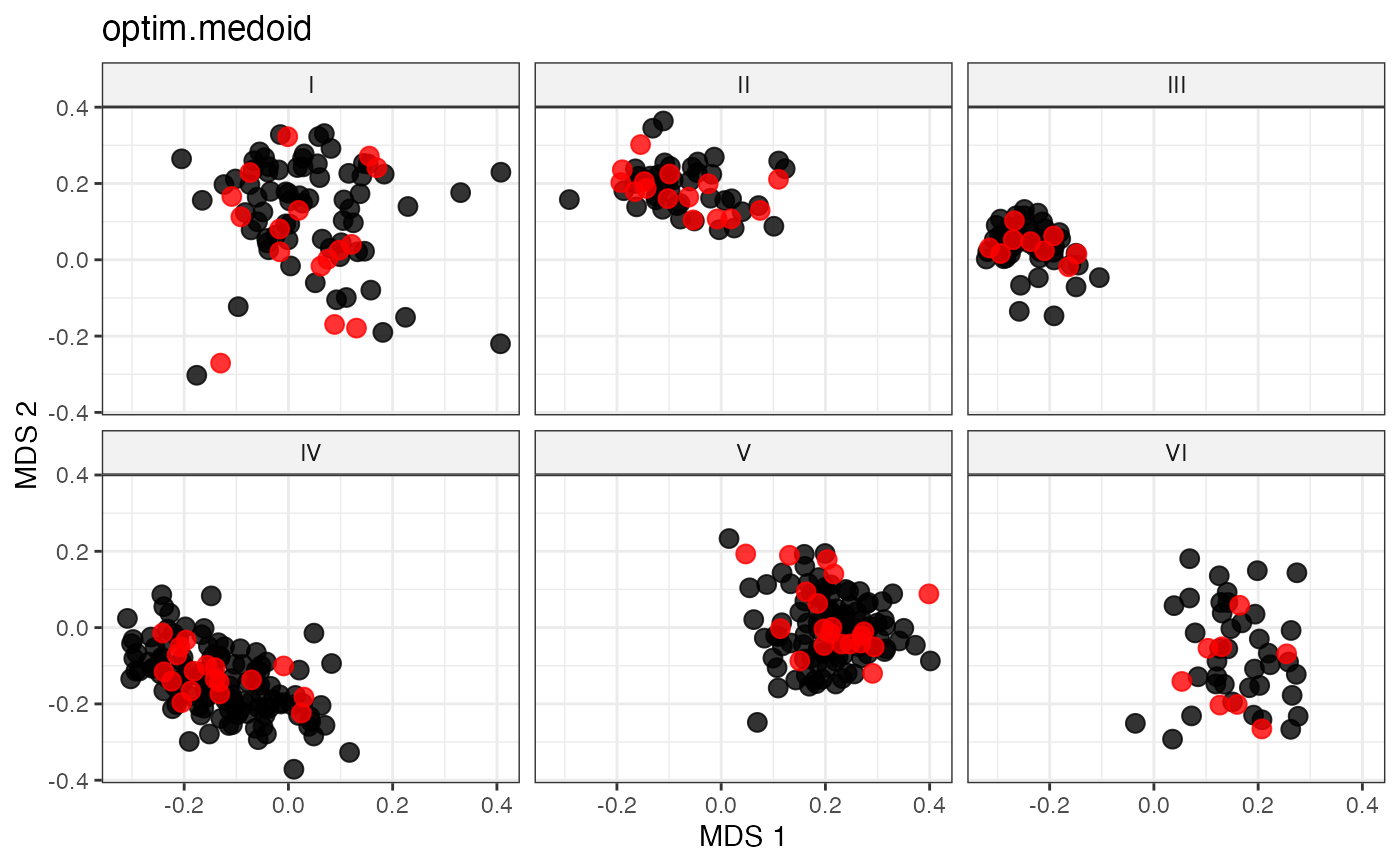
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_pam_out,
use.names = FALSE)) +
labs(title = "optim.medoid")
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Fetch selected accessions by cluster based methods
#~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
# Globally Optimal Medoid Sampling via Partitioning Around Medoids (PAM)
sel_pam_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "optim.medoid")
sel_pam_out
#> $I
#> [1] "TMe-2103" "TMe-28" "TMe-44" "TMe-3419" "TMe-1823" "TMe-952"
#> [7] "TMe-2027" "TMe-882" "TMe-910" "TMe-2944" "TMe-1091" "TMe-566"
#> [13] "TMe-3694" "TMe-3115" "TMe-1425" "TMe-1830"
#>
#> $II
#> [1] "TMe-2021" "TMe-681" "TMe-890" "TMe-2951" "TMe-3557" "TMe-3239"
#> [7] "TMe-1385" "TMe-2352" "TMe-1831" "TMe-1754" "TMe-1668" "TMe-409"
#> [13] "TMe-2033" "TMe-2611" "TMe-3200"
#>
#> $III
#> [1] "TMe-3007" "TMe-2394" "TMe-161" "TMe-2356" "TMe-123" "TMe-3335" "TMe-617"
#> [8] "TMe-2748" "TMe-1790"
#>
#> $IV
#> [1] "TMe-594" "TMe-12" "TMe-3378" "TMe-3409" "TMe-1027" "TMe-1155"
#> [7] "TMe-428" "TMe-170" "TMe-2947" "TMe-1139" "TMe-57" "TMe-2458"
#> [13] "TMe-525" "TMe-3068" "TMe-3265" "TMe-3562" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1979" "TMe-629" "TMe-344" "TMe-1234" "TMe-1788" "TMe-288"
#> [7] "TMe-1160" "TMe-954" "TMe-439" "TMe-245" "TMe-645" "TMe-2907"
#> [13] "TMe-647" "TMe-487" "TMe-723" "TMe-1622" "TMe-1880" "TMe-1559"
#> [19] "TMe-2853" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1608" "TMe-2196" "TMe-936" "TMe-1503" "TMe-1217" "TMe-620"
#> [8] "TMe-1445"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_pam_out,
use.names = FALSE)) +
labs(title = "optim.medoid")
 # Cluster-Based Sampling via K-means (Naes Method)
sel_naes_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "naes")
sel_naes_out
#> $I
#> [1] "TMe-2944" "TMe-569" "TMe-3539" "TMe-2967" "TMe-500" "TMe-28"
#> [7] "TMe-1451" "TMe-2103" "TMe-3252" "TMe-1170" "TMe-2810" "TMe-1091"
#> [13] "TMe-1930" "TMe-3104" "TMe-1532" "TMe-1830"
#>
#> $II
#> [1] "TMe-3284" "TMe-369" "TMe-539" "TMe-681" "TMe-2021" "TMe-1385"
#> [7] "TMe-339" "TMe-890" "TMe-3800" "TMe-3557" "TMe-3366" "TMe-3766"
#> [13] "TMe-3447" "TMe-74" "TMe-3495"
#>
#> $III
#> [1] "TMe-1198" "TMe-70" "TMe-1863" "TMe-3569" "TMe-3556" "TMe-3644" "TMe-3638"
#> [8] "TMe-2374" "TMe-1790"
#>
#> $IV
#> [1] "TMe-170" "TMe-191" "TMe-2947" "TMe-3428" "TMe-2807" "TMe-266"
#> [7] "TMe-241" "TMe-3585" "TMe-3459" "TMe-1330" "TMe-3232" "TMe-3265"
#> [13] "TMe-3581" "TMe-3214" "TMe-1434" "TMe-1336" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1003" "TMe-1401" "TMe-682" "TMe-954" "TMe-627" "TMe-1788"
#> [7] "TMe-344" "TMe-755" "TMe-1268" "TMe-629" "TMe-348" "TMe-419"
#> [13] "TMe-1979" "TMe-167" "TMe-2853" "TMe-745" "TMe-1037" "TMe-288"
#> [19] "TMe-362" "TMe-2018"
#>
#> $VI
#> [1] "TMe-936" "TMe-1062" "TMe-1756" "TMe-1217" "TMe-3177" "TMe-1661" "TMe-693"
#> [8] "TMe-2196"
#>
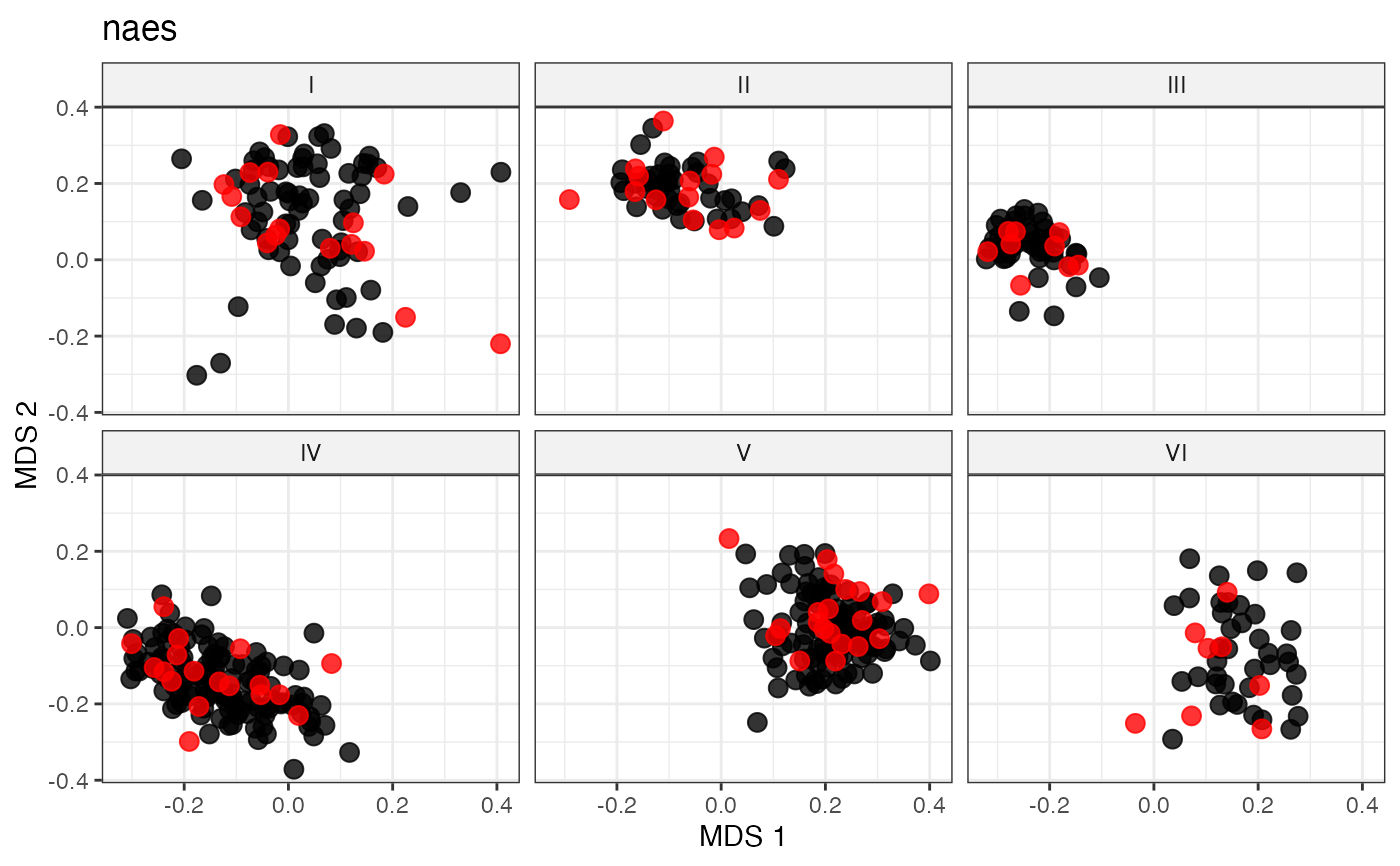
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_naes_out,
use.names = FALSE)) +
labs(title = "naes")
# Cluster-Based Sampling via K-means (Naes Method)
sel_naes_out <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "naes")
sel_naes_out
#> $I
#> [1] "TMe-2944" "TMe-569" "TMe-3539" "TMe-2967" "TMe-500" "TMe-28"
#> [7] "TMe-1451" "TMe-2103" "TMe-3252" "TMe-1170" "TMe-2810" "TMe-1091"
#> [13] "TMe-1930" "TMe-3104" "TMe-1532" "TMe-1830"
#>
#> $II
#> [1] "TMe-3284" "TMe-369" "TMe-539" "TMe-681" "TMe-2021" "TMe-1385"
#> [7] "TMe-339" "TMe-890" "TMe-3800" "TMe-3557" "TMe-3366" "TMe-3766"
#> [13] "TMe-3447" "TMe-74" "TMe-3495"
#>
#> $III
#> [1] "TMe-1198" "TMe-70" "TMe-1863" "TMe-3569" "TMe-3556" "TMe-3644" "TMe-3638"
#> [8] "TMe-2374" "TMe-1790"
#>
#> $IV
#> [1] "TMe-170" "TMe-191" "TMe-2947" "TMe-3428" "TMe-2807" "TMe-266"
#> [7] "TMe-241" "TMe-3585" "TMe-3459" "TMe-1330" "TMe-3232" "TMe-3265"
#> [13] "TMe-3581" "TMe-3214" "TMe-1434" "TMe-1336" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1003" "TMe-1401" "TMe-682" "TMe-954" "TMe-627" "TMe-1788"
#> [7] "TMe-344" "TMe-755" "TMe-1268" "TMe-629" "TMe-348" "TMe-419"
#> [13] "TMe-1979" "TMe-167" "TMe-2853" "TMe-745" "TMe-1037" "TMe-288"
#> [19] "TMe-362" "TMe-2018"
#>
#> $VI
#> [1] "TMe-936" "TMe-1062" "TMe-1756" "TMe-1217" "TMe-3177" "TMe-1661" "TMe-693"
#> [8] "TMe-2196"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_naes_out,
use.names = FALSE)) +
labs(title = "naes")
 # Cluster-Based Sampling via Hierarchical Clustering with Random Selection
## UPGMA
sel_hclust_random_out1 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "average")
sel_hclust_random_out1
#> $I
#> [1] "TMe-888" "TMe-3419" "TMe-2975" "TMe-2943" "TMe-1960" "TMe-2027"
#> [7] "TMe-2785" "TMe-3262" "TMe-2934" "TMe-1190" "TMe-2967" "TMe-3115"
#> [13] "TMe-1425" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2412" "TMe-369" "TMe-890" "TMe-3547" "TMe-196" "TMe-3239"
#> [7] "TMe-1385" "TMe-2352" "TMe-3447" "TMe-2997" "TMe-2033" "TMe-2952"
#> [13] "TMe-2611" "TMe-171" "TMe-3200"
#>
#> $III
#> [1] "TMe-617" "TMe-2270" "TMe-161" "TMe-1819" "TMe-425" "TMe-1176" "TMe-3336"
#> [8] "TMe-3230" "TMe-1790"
#>
#> $IV
#> [1] "TMe-1297" "TMe-3242" "TMe-3409" "TMe-1020" "TMe-1456" "TMe-368"
#> [7] "TMe-460" "TMe-3189" "TMe-3255" "TMe-761" "TMe-2567" "TMe-3273"
#> [13] "TMe-812" "TMe-1123" "TMe-186" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1760" "TMe-688" "TMe-363" "TMe-2750" "TMe-2425" "TMe-877"
#> [7] "TMe-1541" "TMe-362" "TMe-2907" "TMe-1199" "TMe-723" "TMe-755"
#> [13] "TMe-667" "TMe-2853" "TMe-3329" "TMe-712" "TMe-2355" "TMe-823"
#> [19] "TMe-2003" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1302" "TMe-1744" "TMe-1239" "TMe-625" "TMe-1816" "TMe-854" "TMe-1566"
#> [8] "TMe-2983"
#>
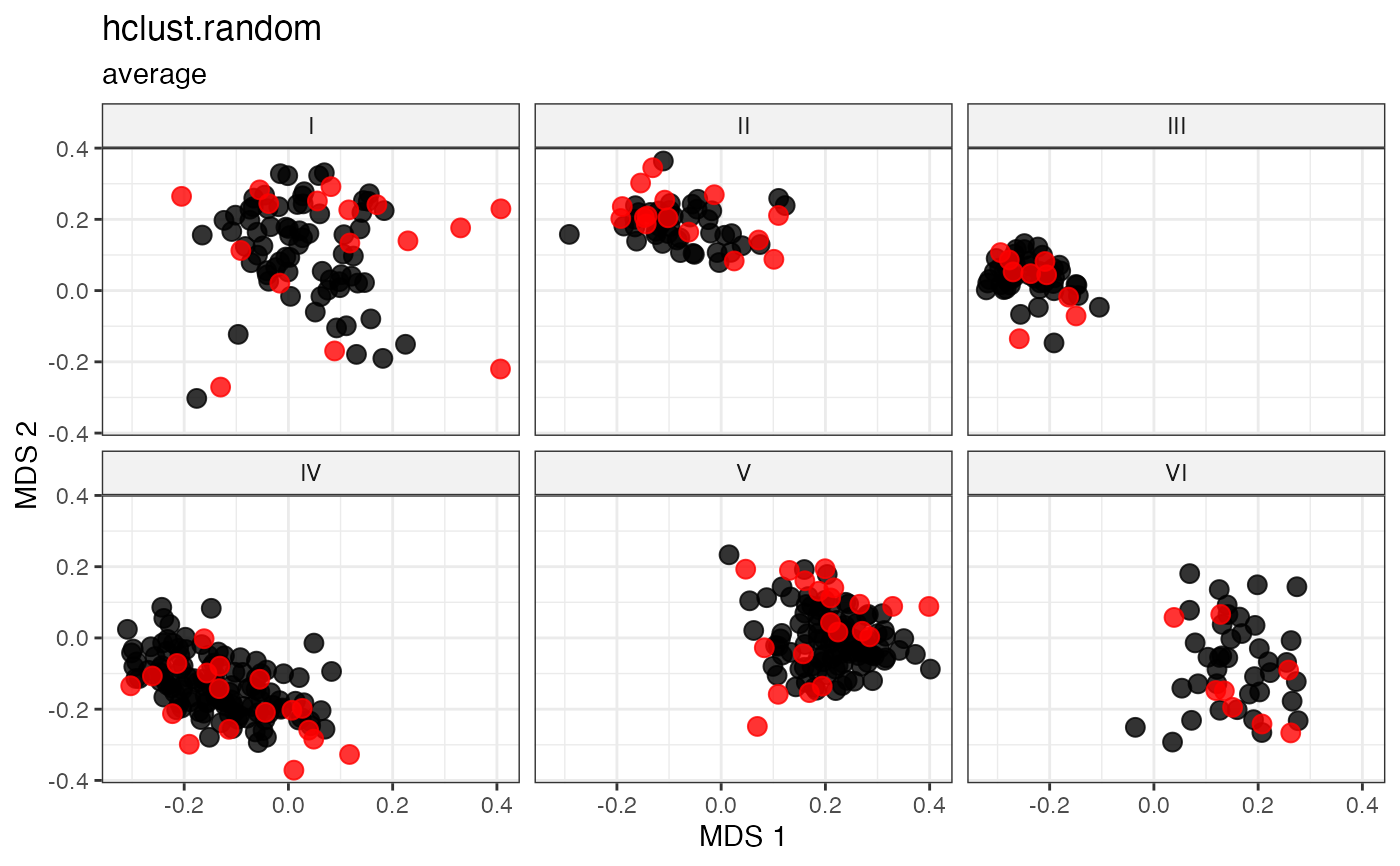
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out1,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "average")
# Cluster-Based Sampling via Hierarchical Clustering with Random Selection
## UPGMA
sel_hclust_random_out1 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "average")
sel_hclust_random_out1
#> $I
#> [1] "TMe-888" "TMe-3419" "TMe-2975" "TMe-2943" "TMe-1960" "TMe-2027"
#> [7] "TMe-2785" "TMe-3262" "TMe-2934" "TMe-1190" "TMe-2967" "TMe-3115"
#> [13] "TMe-1425" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2412" "TMe-369" "TMe-890" "TMe-3547" "TMe-196" "TMe-3239"
#> [7] "TMe-1385" "TMe-2352" "TMe-3447" "TMe-2997" "TMe-2033" "TMe-2952"
#> [13] "TMe-2611" "TMe-171" "TMe-3200"
#>
#> $III
#> [1] "TMe-617" "TMe-2270" "TMe-161" "TMe-1819" "TMe-425" "TMe-1176" "TMe-3336"
#> [8] "TMe-3230" "TMe-1790"
#>
#> $IV
#> [1] "TMe-1297" "TMe-3242" "TMe-3409" "TMe-1020" "TMe-1456" "TMe-368"
#> [7] "TMe-460" "TMe-3189" "TMe-3255" "TMe-761" "TMe-2567" "TMe-3273"
#> [13] "TMe-812" "TMe-1123" "TMe-186" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1760" "TMe-688" "TMe-363" "TMe-2750" "TMe-2425" "TMe-877"
#> [7] "TMe-1541" "TMe-362" "TMe-2907" "TMe-1199" "TMe-723" "TMe-755"
#> [13] "TMe-667" "TMe-2853" "TMe-3329" "TMe-712" "TMe-2355" "TMe-823"
#> [19] "TMe-2003" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1302" "TMe-1744" "TMe-1239" "TMe-625" "TMe-1816" "TMe-854" "TMe-1566"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out1,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "average")
 ## Single-linkage
sel_hclust_random_out2 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "single")
sel_hclust_random_out2
#> $I
#> [1] "TMe-3261" "TMe-3051" "TMe-815" "TMe-2934" "TMe-566" "TMe-3481"
#> [7] "TMe-2967" "TMe-3115" "TMe-1425" "TMe-569" "TMe-3087" "TMe-3112"
#> [13] "TMe-2785" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2412" "TMe-3447" "TMe-674" "TMe-2352" "TMe-2568" "TMe-2903"
#> [7] "TMe-369" "TMe-2033" "TMe-2952" "TMe-3093" "TMe-40" "TMe-3805"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-155" "TMe-1804" "TMe-234" "TMe-2086" "TMe-381" "TMe-3631" "TMe-261"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-27" "TMe-2956" "TMe-1020" "TMe-3390" "TMe-3255" "TMe-761"
#> [7] "TMe-2567" "TMe-3527" "TMe-3273" "TMe-372" "TMe-3542" "TMe-698"
#> [13] "TMe-1988" "TMe-186" "TMe-812" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1500" "TMe-1375" "TMe-344" "TMe-1534" "TMe-1199" "TMe-723"
#> [7] "TMe-2290" "TMe-667" "TMe-2853" "TMe-3329" "TMe-712" "TMe-362"
#> [13] "TMe-2355" "TMe-419" "TMe-2003" "TMe-730" "TMe-98" "TMe-2907"
#> [19] "TMe-2425" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1676" "TMe-1403" "TMe-751" "TMe-1646" "TMe-1124" "TMe-310" "TMe-598"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out2,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "single")
## Single-linkage
sel_hclust_random_out2 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "single")
sel_hclust_random_out2
#> $I
#> [1] "TMe-3261" "TMe-3051" "TMe-815" "TMe-2934" "TMe-566" "TMe-3481"
#> [7] "TMe-2967" "TMe-3115" "TMe-1425" "TMe-569" "TMe-3087" "TMe-3112"
#> [13] "TMe-2785" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2412" "TMe-3447" "TMe-674" "TMe-2352" "TMe-2568" "TMe-2903"
#> [7] "TMe-369" "TMe-2033" "TMe-2952" "TMe-3093" "TMe-40" "TMe-3805"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-155" "TMe-1804" "TMe-234" "TMe-2086" "TMe-381" "TMe-3631" "TMe-261"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-27" "TMe-2956" "TMe-1020" "TMe-3390" "TMe-3255" "TMe-761"
#> [7] "TMe-2567" "TMe-3527" "TMe-3273" "TMe-372" "TMe-3542" "TMe-698"
#> [13] "TMe-1988" "TMe-186" "TMe-812" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1500" "TMe-1375" "TMe-344" "TMe-1534" "TMe-1199" "TMe-723"
#> [7] "TMe-2290" "TMe-667" "TMe-2853" "TMe-3329" "TMe-712" "TMe-362"
#> [13] "TMe-2355" "TMe-419" "TMe-2003" "TMe-730" "TMe-98" "TMe-2907"
#> [19] "TMe-2425" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1676" "TMe-1403" "TMe-751" "TMe-1646" "TMe-1124" "TMe-310" "TMe-598"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out2,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "single")
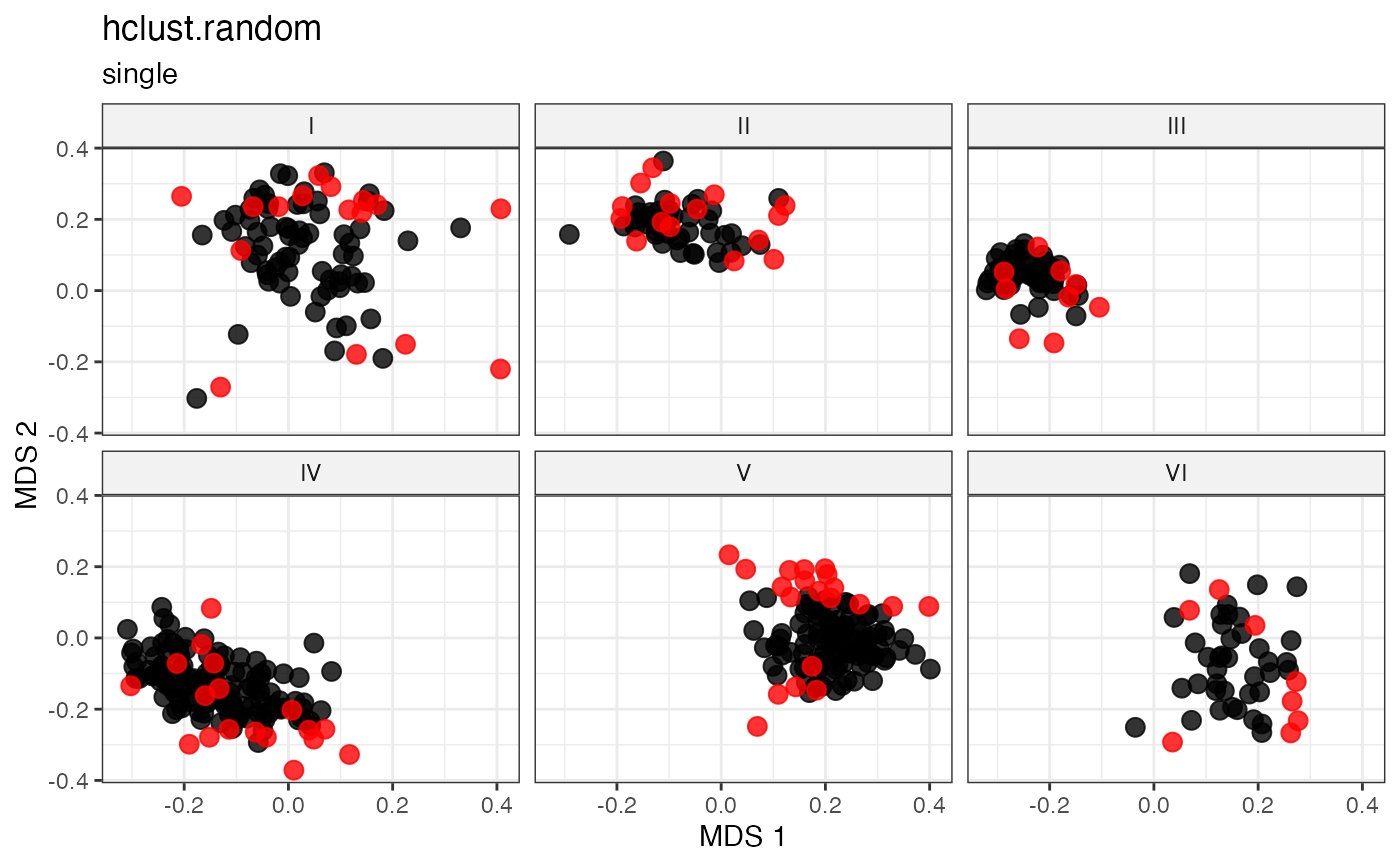 ## Complete-linkage
sel_hclust_random_out3 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "complete")
sel_hclust_random_out3
#> $I
#> [1] "TMe-2513" "TMe-3051" "TMe-2810" "TMe-3252" "TMe-1914" "TMe-306"
#> [7] "TMe-500" "TMe-1096" "TMe-2785" "TMe-3262" "TMe-3115" "TMe-566"
#> [13] "TMe-2943" "TMe-1218" "TMe-1425" "TMe-1830"
#>
#> $II
#> [1] "TMe-796" "TMe-2757" "TMe-3093" "TMe-3258" "TMe-1732" "TMe-3495"
#> [7] "TMe-74" "TMe-171" "TMe-3766" "TMe-1831" "TMe-3366" "TMe-455"
#> [13] "TMe-3805" "TMe-2952" "TMe-1698"
#>
#> $III
#> [1] "TMe-1787" "TMe-3638" "TMe-2374" "TMe-3644" "TMe-1230" "TMe-3620" "TMe-1738"
#> [8] "TMe-2748" "TMe-1790"
#>
#> $IV
#> [1] "TMe-259" "TMe-2958" "TMe-3537" "TMe-3409" "TMe-1297" "TMe-1336"
#> [7] "TMe-3378" "TMe-3558" "TMe-1456" "TMe-57" "TMe-1700" "TMe-266"
#> [13] "TMe-460" "TMe-761" "TMe-1434" "TMe-3581" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-723" "TMe-795" "TMe-439" "TMe-363" "TMe-1440" "TMe-920"
#> [7] "TMe-98" "TMe-877" "TMe-1160" "TMe-1541" "TMe-651" "TMe-645"
#> [13] "TMe-816" "TMe-2907" "TMe-1414" "TMe-419" "TMe-2853" "TMe-3329"
#> [19] "TMe-623" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1744" "TMe-1816" "TMe-968" "TMe-531" "TMe-620" "TMe-1392"
#> [8] "TMe-1992"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out3,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "complete")
## Complete-linkage
sel_hclust_random_out3 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "complete")
sel_hclust_random_out3
#> $I
#> [1] "TMe-2513" "TMe-3051" "TMe-2810" "TMe-3252" "TMe-1914" "TMe-306"
#> [7] "TMe-500" "TMe-1096" "TMe-2785" "TMe-3262" "TMe-3115" "TMe-566"
#> [13] "TMe-2943" "TMe-1218" "TMe-1425" "TMe-1830"
#>
#> $II
#> [1] "TMe-796" "TMe-2757" "TMe-3093" "TMe-3258" "TMe-1732" "TMe-3495"
#> [7] "TMe-74" "TMe-171" "TMe-3766" "TMe-1831" "TMe-3366" "TMe-455"
#> [13] "TMe-3805" "TMe-2952" "TMe-1698"
#>
#> $III
#> [1] "TMe-1787" "TMe-3638" "TMe-2374" "TMe-3644" "TMe-1230" "TMe-3620" "TMe-1738"
#> [8] "TMe-2748" "TMe-1790"
#>
#> $IV
#> [1] "TMe-259" "TMe-2958" "TMe-3537" "TMe-3409" "TMe-1297" "TMe-1336"
#> [7] "TMe-3378" "TMe-3558" "TMe-1456" "TMe-57" "TMe-1700" "TMe-266"
#> [13] "TMe-460" "TMe-761" "TMe-1434" "TMe-3581" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-723" "TMe-795" "TMe-439" "TMe-363" "TMe-1440" "TMe-920"
#> [7] "TMe-98" "TMe-877" "TMe-1160" "TMe-1541" "TMe-651" "TMe-645"
#> [13] "TMe-816" "TMe-2907" "TMe-1414" "TMe-419" "TMe-2853" "TMe-3329"
#> [19] "TMe-623" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1744" "TMe-1816" "TMe-968" "TMe-531" "TMe-620" "TMe-1392"
#> [8] "TMe-1992"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out3,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "complete")
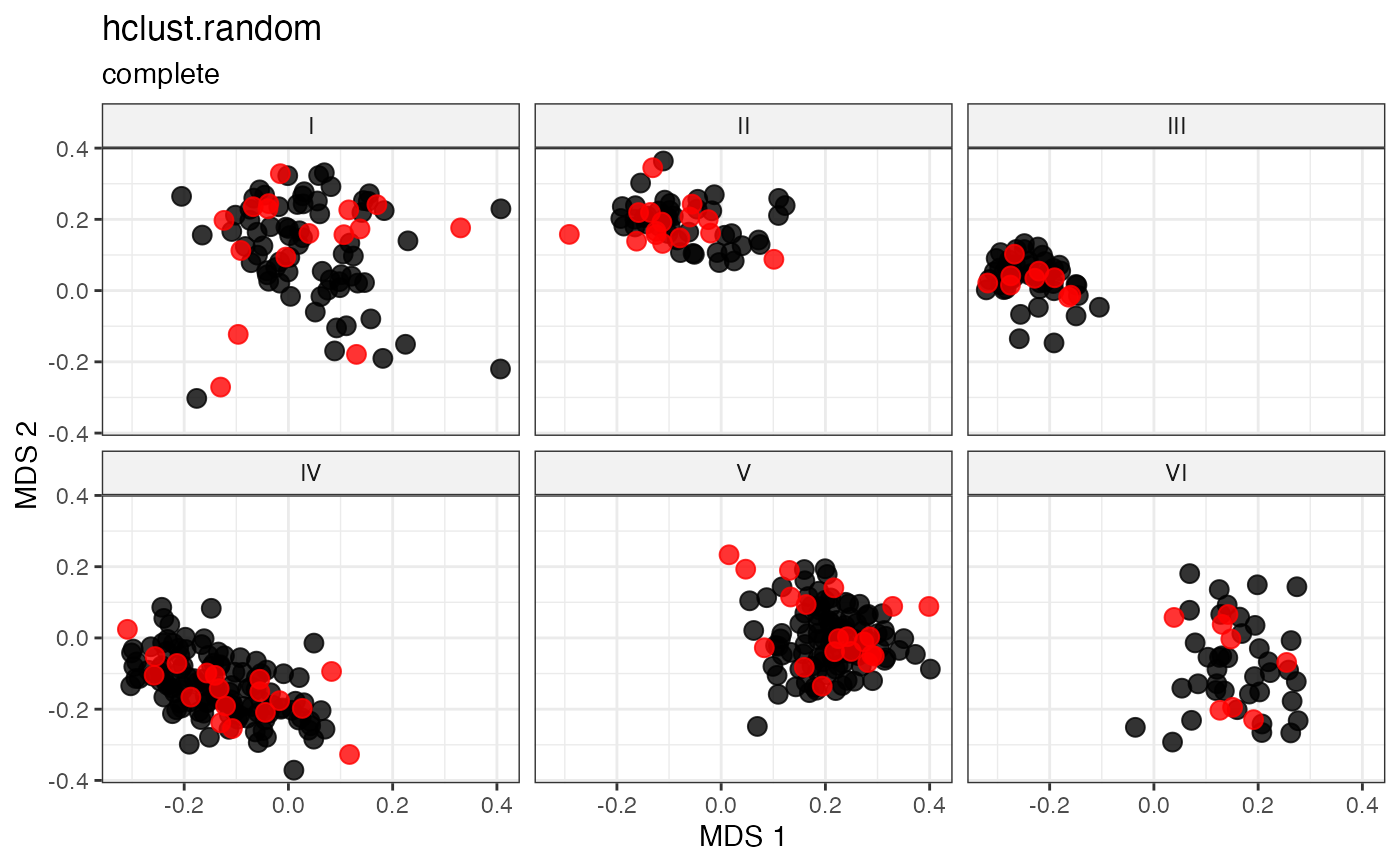 ## Ward's D
sel_hclust_random_out4 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "ward.D")
sel_hclust_random_out4
#> $I
#> [1] "TMe-2152" "TMe-3719" "TMe-3236" "TMe-3685" "TMe-1581" "TMe-1117"
#> [7] "TMe-500" "TMe-1091" "TMe-3110" "TMe-1914" "TMe-2944" "TMe-569"
#> [13] "TMe-1096" "TMe-778" "TMe-3115" "TMe-1830"
#>
#> $II
#> [1] "TMe-2000" "TMe-681" "TMe-455" "TMe-3547" "TMe-3557" "TMe-2997"
#> [7] "TMe-74" "TMe-1385" "TMe-2352" "TMe-1831" "TMe-171" "TMe-2568"
#> [13] "TMe-2329" "TMe-1474" "TMe-3530"
#>
#> $III
#> [1] "TMe-3638" "TMe-1198" "TMe-2968" "TMe-3088" "TMe-381" "TMe-1230" "TMe-3133"
#> [8] "TMe-155" "TMe-1790"
#>
#> $IV
#> [1] "TMe-3103" "TMe-2841" "TMe-875" "TMe-2956" "TMe-761" "TMe-427"
#> [7] "TMe-65" "TMe-897" "TMe-2240" "TMe-1579" "TMe-3218" "TMe-1167"
#> [13] "TMe-2946" "TMe-108" "TMe-12" "TMe-1434" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1979" "TMe-623" "TMe-224" "TMe-543" "TMe-2750" "TMe-2425"
#> [7] "TMe-877" "TMe-1160" "TMe-1534" "TMe-362" "TMe-288" "TMe-707"
#> [13] "TMe-627" "TMe-1880" "TMe-2192" "TMe-1760" "TMe-667" "TMe-2355"
#> [19] "TMe-348" "TMe-2018"
#>
#> $VI
#> [1] "TMe-631" "TMe-1608" "TMe-1124" "TMe-1676" "TMe-1592" "TMe-1035" "TMe-1403"
#> [8] "TMe-1445"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out4,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "ward.D")
## Ward's D
sel_hclust_random_out4 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "ward.D")
sel_hclust_random_out4
#> $I
#> [1] "TMe-2152" "TMe-3719" "TMe-3236" "TMe-3685" "TMe-1581" "TMe-1117"
#> [7] "TMe-500" "TMe-1091" "TMe-3110" "TMe-1914" "TMe-2944" "TMe-569"
#> [13] "TMe-1096" "TMe-778" "TMe-3115" "TMe-1830"
#>
#> $II
#> [1] "TMe-2000" "TMe-681" "TMe-455" "TMe-3547" "TMe-3557" "TMe-2997"
#> [7] "TMe-74" "TMe-1385" "TMe-2352" "TMe-1831" "TMe-171" "TMe-2568"
#> [13] "TMe-2329" "TMe-1474" "TMe-3530"
#>
#> $III
#> [1] "TMe-3638" "TMe-1198" "TMe-2968" "TMe-3088" "TMe-381" "TMe-1230" "TMe-3133"
#> [8] "TMe-155" "TMe-1790"
#>
#> $IV
#> [1] "TMe-3103" "TMe-2841" "TMe-875" "TMe-2956" "TMe-761" "TMe-427"
#> [7] "TMe-65" "TMe-897" "TMe-2240" "TMe-1579" "TMe-3218" "TMe-1167"
#> [13] "TMe-2946" "TMe-108" "TMe-12" "TMe-1434" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1979" "TMe-623" "TMe-224" "TMe-543" "TMe-2750" "TMe-2425"
#> [7] "TMe-877" "TMe-1160" "TMe-1534" "TMe-362" "TMe-288" "TMe-707"
#> [13] "TMe-627" "TMe-1880" "TMe-2192" "TMe-1760" "TMe-667" "TMe-2355"
#> [19] "TMe-348" "TMe-2018"
#>
#> $VI
#> [1] "TMe-631" "TMe-1608" "TMe-1124" "TMe-1676" "TMe-1592" "TMe-1035" "TMe-1403"
#> [8] "TMe-1445"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out4,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "ward.D")
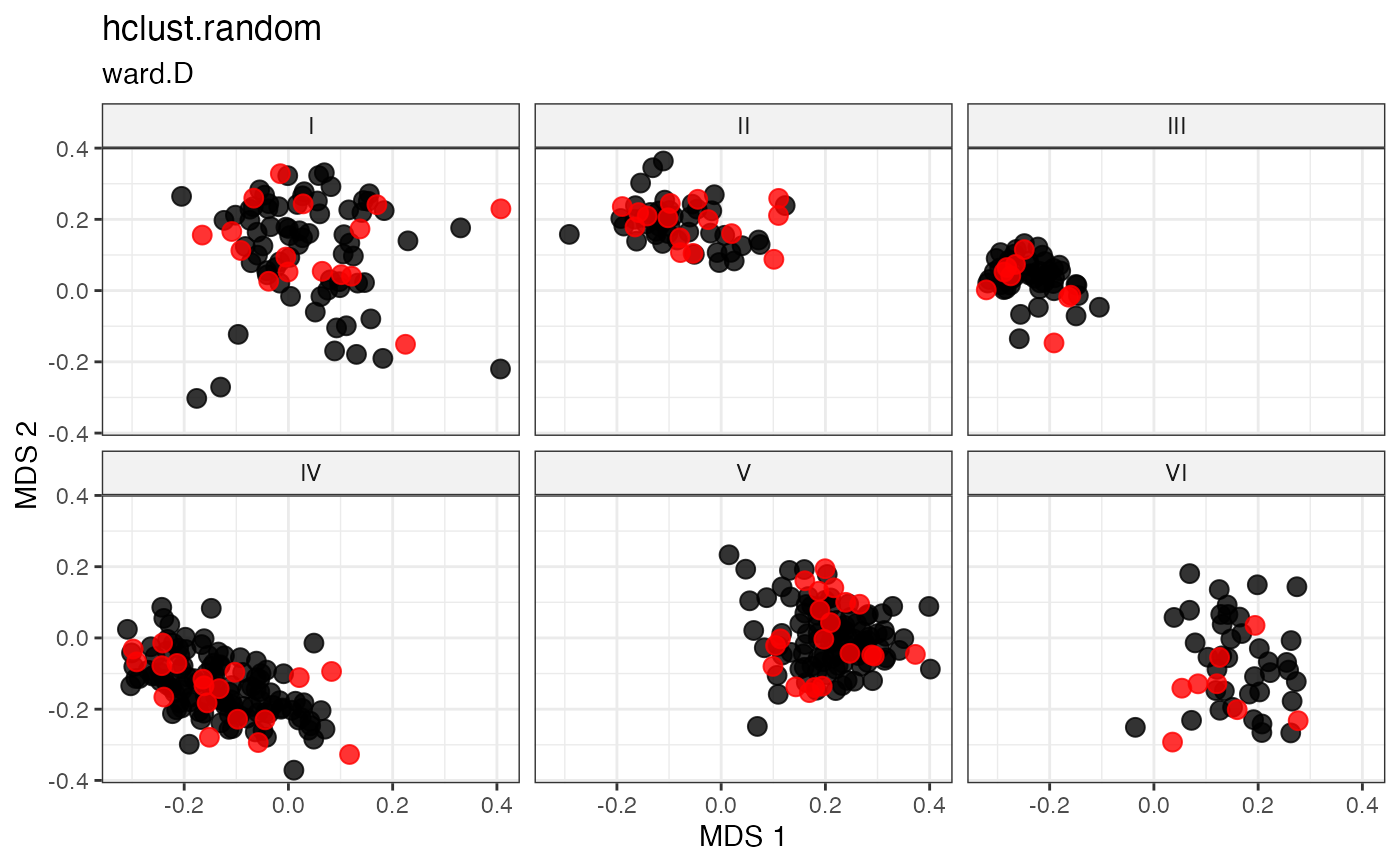 ## WPGMA
sel_hclust_random_out5 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "mcquitty")
sel_hclust_random_out5
#> $I
#> [1] "TMe-937" "TMe-3051" "TMe-888" "TMe-3087" "TMe-952" "TMe-500"
#> [7] "TMe-3111" "TMe-2943" "TMe-815" "TMe-566" "TMe-2810" "TMe-2967"
#> [13] "TMe-1425" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2021" "TMe-2257" "TMe-3800" "TMe-3258" "TMe-3557" "TMe-339"
#> [7] "TMe-1385" "TMe-3766" "TMe-3530" "TMe-1668" "TMe-409" "TMe-2033"
#> [13] "TMe-2952" "TMe-171" "TMe-3200"
#>
#> $III
#> [1] "TMe-3207" "TMe-2502" "TMe-161" "TMe-2356" "TMe-3100" "TMe-3631" "TMe-3397"
#> [8] "TMe-2968" "TMe-1790"
#>
#> $IV
#> [1] "TMe-386" "TMe-3562" "TMe-2367" "TMe-3585" "TMe-1020" "TMe-1456"
#> [7] "TMe-956" "TMe-25" "TMe-3255" "TMe-761" "TMe-2567" "TMe-3273"
#> [13] "TMe-812" "TMe-2928" "TMe-186" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1934" "TMe-1101" "TMe-344" "TMe-362" "TMe-1427" "TMe-2425"
#> [7] "TMe-2016" "TMe-1534" "TMe-682" "TMe-647" "TMe-1500" "TMe-723"
#> [13] "TMe-730" "TMe-1694" "TMe-419" "TMe-2853" "TMe-3329" "TMe-2355"
#> [19] "TMe-2003" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1062" "TMe-854" "TMe-1239" "TMe-1945" "TMe-1035" "TMe-1445" "TMe-1566"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out5,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "mcquitty")
## WPGMA
sel_hclust_random_out5 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "mcquitty")
sel_hclust_random_out5
#> $I
#> [1] "TMe-937" "TMe-3051" "TMe-888" "TMe-3087" "TMe-952" "TMe-500"
#> [7] "TMe-3111" "TMe-2943" "TMe-815" "TMe-566" "TMe-2810" "TMe-2967"
#> [13] "TMe-1425" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2021" "TMe-2257" "TMe-3800" "TMe-3258" "TMe-3557" "TMe-339"
#> [7] "TMe-1385" "TMe-3766" "TMe-3530" "TMe-1668" "TMe-409" "TMe-2033"
#> [13] "TMe-2952" "TMe-171" "TMe-3200"
#>
#> $III
#> [1] "TMe-3207" "TMe-2502" "TMe-161" "TMe-2356" "TMe-3100" "TMe-3631" "TMe-3397"
#> [8] "TMe-2968" "TMe-1790"
#>
#> $IV
#> [1] "TMe-386" "TMe-3562" "TMe-2367" "TMe-3585" "TMe-1020" "TMe-1456"
#> [7] "TMe-956" "TMe-25" "TMe-3255" "TMe-761" "TMe-2567" "TMe-3273"
#> [13] "TMe-812" "TMe-2928" "TMe-186" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1934" "TMe-1101" "TMe-344" "TMe-362" "TMe-1427" "TMe-2425"
#> [7] "TMe-2016" "TMe-1534" "TMe-682" "TMe-647" "TMe-1500" "TMe-723"
#> [13] "TMe-730" "TMe-1694" "TMe-419" "TMe-2853" "TMe-3329" "TMe-2355"
#> [19] "TMe-2003" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1062" "TMe-854" "TMe-1239" "TMe-1945" "TMe-1035" "TMe-1445" "TMe-1566"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out5,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "mcquitty")
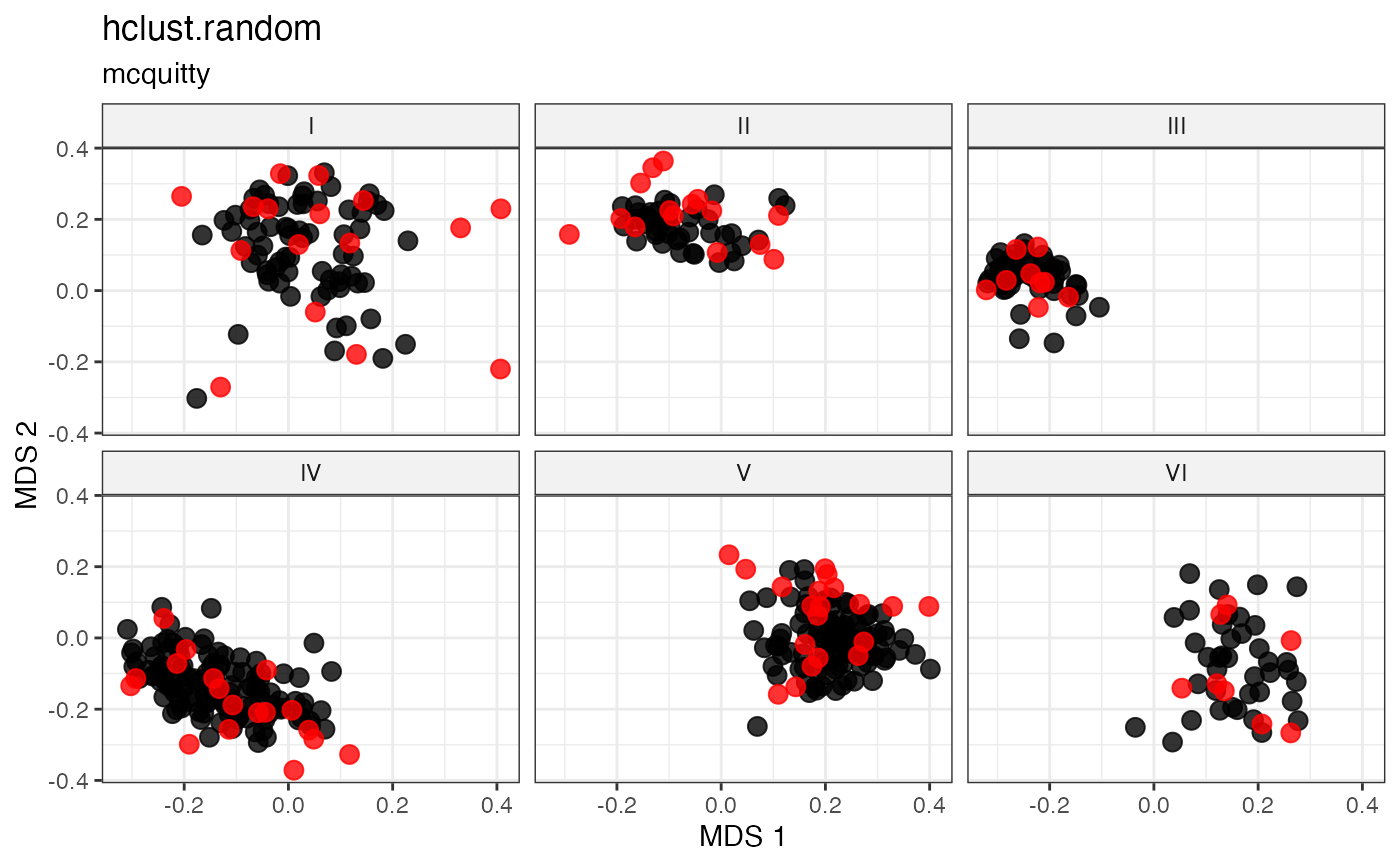 ## WPGMC
sel_hclust_random_out6 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "median")
sel_hclust_random_out6
#> $I
#> [1] "TMe-3261" "TMe-3051" "TMe-815" "TMe-2934" "TMe-3262" "TMe-2943"
#> [7] "TMe-2967" "TMe-1096" "TMe-1425" "TMe-569" "TMe-3112" "TMe-2785"
#> [13] "TMe-3437" "TMe-3111" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-3495" "TMe-3800" "TMe-674" "TMe-2352" "TMe-3766" "TMe-2329"
#> [7] "TMe-369" "TMe-2033" "TMe-2952" "TMe-3093" "TMe-3805" "TMe-3547"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-1787" "TMe-148" "TMe-3100" "TMe-3631" "TMe-14" "TMe-261" "TMe-3397"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-3537" "TMe-2956" "TMe-1020" "TMe-1376" "TMe-3255" "TMe-761"
#> [7] "TMe-3527" "TMe-3273" "TMe-3189" "TMe-698" "TMe-1434" "TMe-427"
#> [13] "TMe-2971" "TMe-1988" "TMe-186" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2138" "TMe-688" "TMe-769" "TMe-2151" "TMe-256" "TMe-723"
#> [7] "TMe-1880" "TMe-2290" "TMe-2853" "TMe-3329" "TMe-423" "TMe-712"
#> [13] "TMe-362" "TMe-2355" "TMe-419" "TMe-1934" "TMe-2003" "TMe-2425"
#> [19] "TMe-2855" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1445" "TMe-693" "TMe-751" "TMe-1646" "TMe-1124" "TMe-1661" "TMe-2791"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out6,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "median")
## WPGMC
sel_hclust_random_out6 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "median")
sel_hclust_random_out6
#> $I
#> [1] "TMe-3261" "TMe-3051" "TMe-815" "TMe-2934" "TMe-3262" "TMe-2943"
#> [7] "TMe-2967" "TMe-1096" "TMe-1425" "TMe-569" "TMe-3112" "TMe-2785"
#> [13] "TMe-3437" "TMe-3111" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-3495" "TMe-3800" "TMe-674" "TMe-2352" "TMe-3766" "TMe-2329"
#> [7] "TMe-369" "TMe-2033" "TMe-2952" "TMe-3093" "TMe-3805" "TMe-3547"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-1787" "TMe-148" "TMe-3100" "TMe-3631" "TMe-14" "TMe-261" "TMe-3397"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-3537" "TMe-2956" "TMe-1020" "TMe-1376" "TMe-3255" "TMe-761"
#> [7] "TMe-3527" "TMe-3273" "TMe-3189" "TMe-698" "TMe-1434" "TMe-427"
#> [13] "TMe-2971" "TMe-1988" "TMe-186" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2138" "TMe-688" "TMe-769" "TMe-2151" "TMe-256" "TMe-723"
#> [7] "TMe-1880" "TMe-2290" "TMe-2853" "TMe-3329" "TMe-423" "TMe-712"
#> [13] "TMe-362" "TMe-2355" "TMe-419" "TMe-1934" "TMe-2003" "TMe-2425"
#> [19] "TMe-2855" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1445" "TMe-693" "TMe-751" "TMe-1646" "TMe-1124" "TMe-1661" "TMe-2791"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out6,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "median")
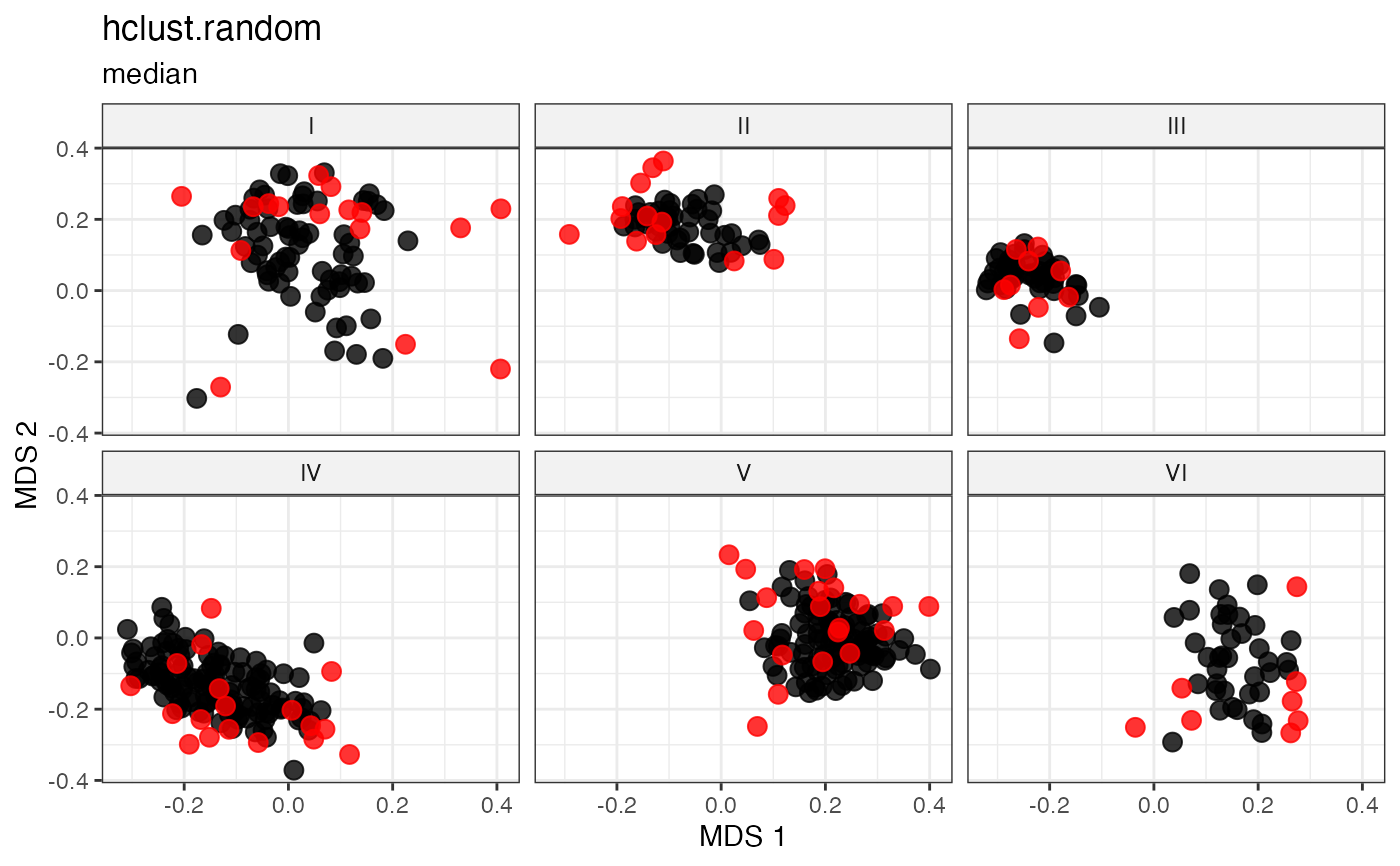 ## UPGMC
sel_hclust_random_out7 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "centroid")
sel_hclust_random_out7
#> $I
#> [1] "TMe-1451" "TMe-500" "TMe-815" "TMe-2934" "TMe-566" "TMe-2943"
#> [7] "TMe-1218" "TMe-2967" "TMe-1190" "TMe-1425" "TMe-569" "TMe-41"
#> [13] "TMe-3437" "TMe-2027" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-1505" "TMe-3800" "TMe-674" "TMe-2352" "TMe-2568" "TMe-3766"
#> [7] "TMe-2329" "TMe-369" "TMe-2033" "TMe-2952" "TMe-1754" "TMe-3805"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-1819" "TMe-234" "TMe-381" "TMe-3631" "TMe-2374" "TMe-3715" "TMe-261"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-18" "TMe-2956" "TMe-1020" "TMe-1376" "TMe-761" "TMe-2567"
#> [7] "TMe-3527" "TMe-3273" "TMe-3581" "TMe-27" "TMe-698" "TMe-1434"
#> [13] "TMe-427" "TMe-1988" "TMe-812" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2213" "TMe-1375" "TMe-344" "TMe-532" "TMe-723" "TMe-2290"
#> [7] "TMe-667" "TMe-224" "TMe-2853" "TMe-3329" "TMe-423" "TMe-712"
#> [13] "TMe-2355" "TMe-419" "TMe-2003" "TMe-627" "TMe-98" "TMe-2750"
#> [19] "TMe-2907" "TMe-2018"
#>
#> $VI
#> [1] "TMe-625" "TMe-693" "TMe-751" "TMe-1124" "TMe-2791" "TMe-1035" "TMe-1992"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out7,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "centroid")
## UPGMC
sel_hclust_random_out7 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "centroid")
sel_hclust_random_out7
#> $I
#> [1] "TMe-1451" "TMe-500" "TMe-815" "TMe-2934" "TMe-566" "TMe-2943"
#> [7] "TMe-1218" "TMe-2967" "TMe-1190" "TMe-1425" "TMe-569" "TMe-41"
#> [13] "TMe-3437" "TMe-2027" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-1505" "TMe-3800" "TMe-674" "TMe-2352" "TMe-2568" "TMe-3766"
#> [7] "TMe-2329" "TMe-369" "TMe-2033" "TMe-2952" "TMe-1754" "TMe-3805"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-1819" "TMe-234" "TMe-381" "TMe-3631" "TMe-2374" "TMe-3715" "TMe-261"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-18" "TMe-2956" "TMe-1020" "TMe-1376" "TMe-761" "TMe-2567"
#> [7] "TMe-3527" "TMe-3273" "TMe-3581" "TMe-27" "TMe-698" "TMe-1434"
#> [13] "TMe-427" "TMe-1988" "TMe-812" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-2213" "TMe-1375" "TMe-344" "TMe-532" "TMe-723" "TMe-2290"
#> [7] "TMe-667" "TMe-224" "TMe-2853" "TMe-3329" "TMe-423" "TMe-712"
#> [13] "TMe-2355" "TMe-419" "TMe-2003" "TMe-627" "TMe-98" "TMe-2750"
#> [19] "TMe-2907" "TMe-2018"
#>
#> $VI
#> [1] "TMe-625" "TMe-693" "TMe-751" "TMe-1124" "TMe-2791" "TMe-1035" "TMe-1992"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out7,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "centroid")
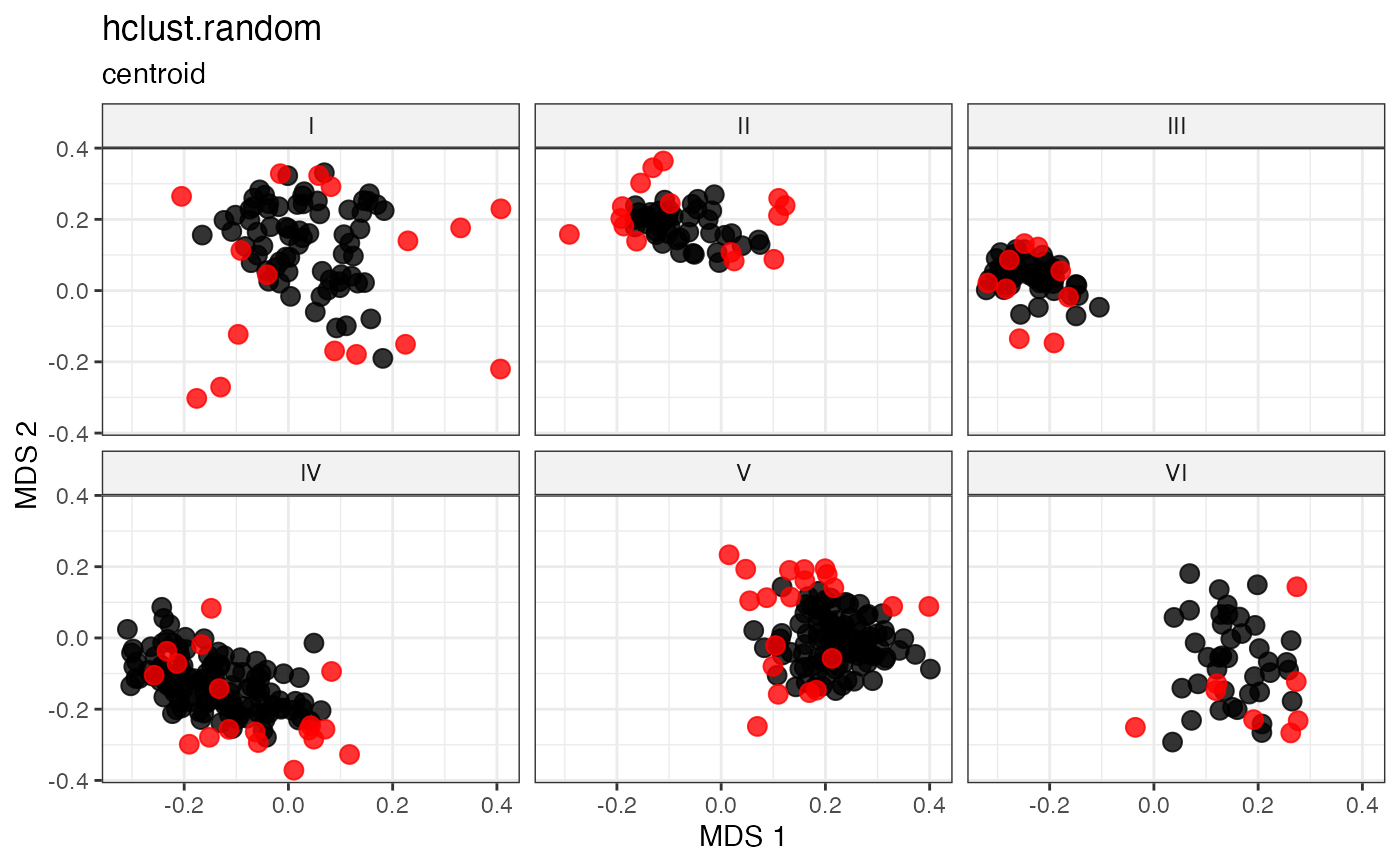 ## Ward's D2
sel_hclust_random_out8 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "ward.D2")
sel_hclust_random_out8
#> $I
#> [1] "TMe-2066" "TMe-3481" "TMe-2027" "TMe-2934" "TMe-3726" "TMe-1589"
#> [7] "TMe-469" "TMe-3142" "TMe-3465" "TMe-2237" "TMe-2944" "TMe-3514"
#> [13] "TMe-3694" "TMe-778" "TMe-3398" "TMe-1830"
#>
#> $II
#> [1] "TMe-2000" "TMe-2257" "TMe-3800" "TMe-3547" "TMe-1505" "TMe-40"
#> [7] "TMe-3200" "TMe-674" "TMe-3766" "TMe-3447" "TMe-171" "TMe-2568"
#> [13] "TMe-1474" "TMe-2033" "TMe-3284"
#>
#> $III
#> [1] "TMe-3715" "TMe-2502" "TMe-2374" "TMe-830" "TMe-425" "TMe-2394" "TMe-2086"
#> [8] "TMe-617" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2971" "TMe-3542" "TMe-875" "TMe-2240" "TMe-1155" "TMe-1336"
#> [7] "TMe-1489" "TMe-3527" "TMe-1246" "TMe-3255" "TMe-266" "TMe-3072"
#> [13] "TMe-241" "TMe-427" "TMe-182" "TMe-2928" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-623" "TMe-323" "TMe-892" "TMe-2138" "TMe-1375" "TMe-769"
#> [7] "TMe-1159" "TMe-745" "TMe-167" "TMe-1293" "TMe-1265" "TMe-707"
#> [13] "TMe-2907" "TMe-1880" "TMe-1003" "TMe-1295" "TMe-419" "TMe-2853"
#> [19] "TMe-98" "TMe-2018"
#>
#> $VI
#> [1] "TMe-505" "TMe-1744" "TMe-1239" "TMe-1076" "TMe-1756" "TMe-1413" "TMe-1217"
#> [8] "TMe-1403"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out8,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "ward.D2")
## Ward's D2
sel_hclust_random_out8 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.random",
hclust.method = "ward.D2")
sel_hclust_random_out8
#> $I
#> [1] "TMe-2066" "TMe-3481" "TMe-2027" "TMe-2934" "TMe-3726" "TMe-1589"
#> [7] "TMe-469" "TMe-3142" "TMe-3465" "TMe-2237" "TMe-2944" "TMe-3514"
#> [13] "TMe-3694" "TMe-778" "TMe-3398" "TMe-1830"
#>
#> $II
#> [1] "TMe-2000" "TMe-2257" "TMe-3800" "TMe-3547" "TMe-1505" "TMe-40"
#> [7] "TMe-3200" "TMe-674" "TMe-3766" "TMe-3447" "TMe-171" "TMe-2568"
#> [13] "TMe-1474" "TMe-2033" "TMe-3284"
#>
#> $III
#> [1] "TMe-3715" "TMe-2502" "TMe-2374" "TMe-830" "TMe-425" "TMe-2394" "TMe-2086"
#> [8] "TMe-617" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2971" "TMe-3542" "TMe-875" "TMe-2240" "TMe-1155" "TMe-1336"
#> [7] "TMe-1489" "TMe-3527" "TMe-1246" "TMe-3255" "TMe-266" "TMe-3072"
#> [13] "TMe-241" "TMe-427" "TMe-182" "TMe-2928" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-623" "TMe-323" "TMe-892" "TMe-2138" "TMe-1375" "TMe-769"
#> [7] "TMe-1159" "TMe-745" "TMe-167" "TMe-1293" "TMe-1265" "TMe-707"
#> [13] "TMe-2907" "TMe-1880" "TMe-1003" "TMe-1295" "TMe-419" "TMe-2853"
#> [19] "TMe-98" "TMe-2018"
#>
#> $VI
#> [1] "TMe-505" "TMe-1744" "TMe-1239" "TMe-1076" "TMe-1756" "TMe-1413" "TMe-1217"
#> [8] "TMe-1403"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_random_out8,
use.names = FALSE)) +
labs(title = "hclust.random", subtitle = "ward.D2")
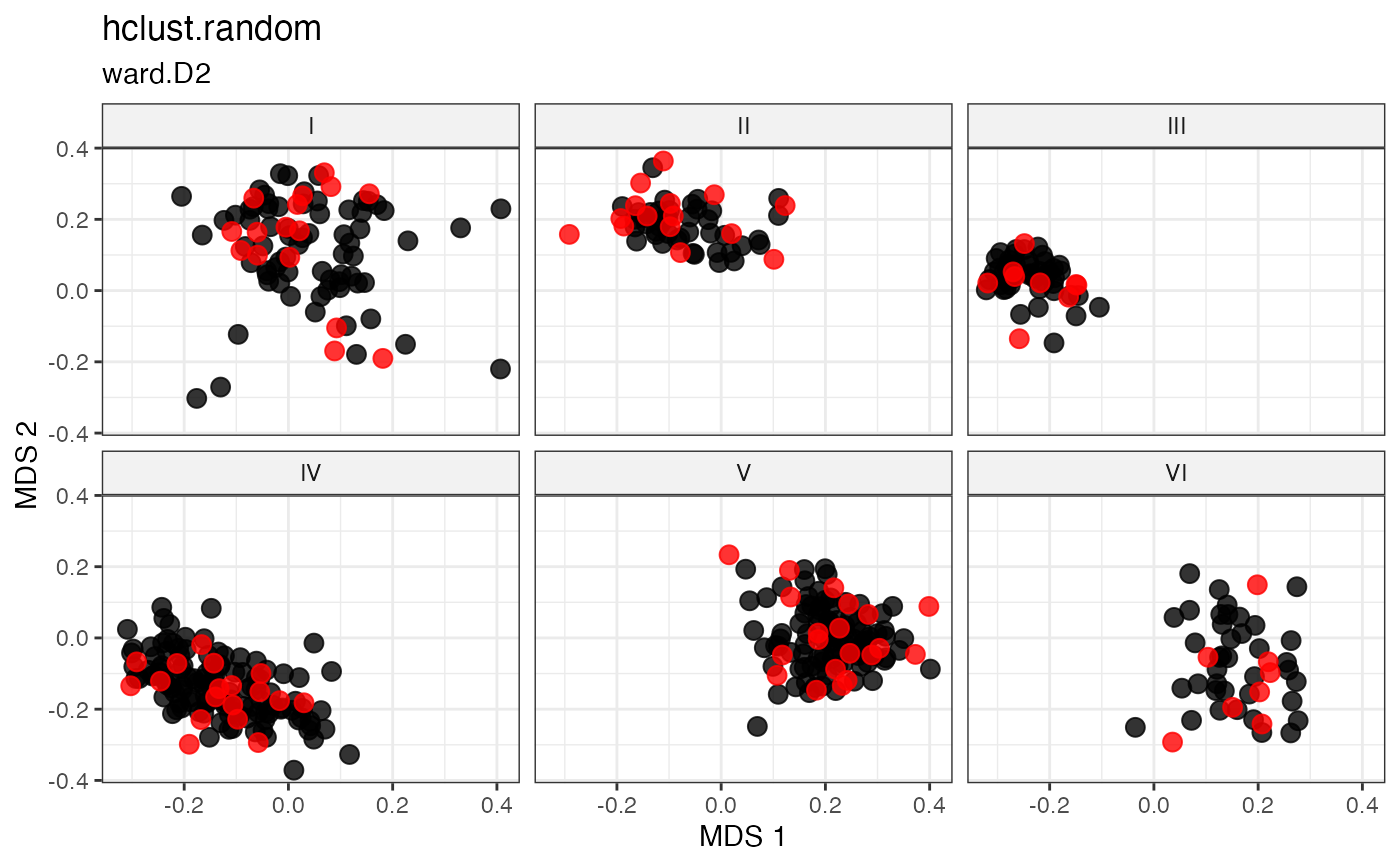 # Cluster-Based Sampling via Hierarchical Clustering with Medoid Selection
## UPGMA
sel_hclust_medoid_out1 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "average")
sel_hclust_medoid_out1
#> $I
#> [1] "TMe-1823" "TMe-3132" "TMe-44" "TMe-3539" "TMe-952" "TMe-500"
#> [7] "TMe-3110" "TMe-815" "TMe-2934" "TMe-566" "TMe-2967" "TMe-3398"
#> [13] "TMe-1425" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2258" "TMe-681" "TMe-890" "TMe-2951" "TMe-3557" "TMe-3239"
#> [7] "TMe-674" "TMe-2352" "TMe-1831" "TMe-3101" "TMe-2033" "TMe-2952"
#> [13] "TMe-3284" "TMe-171" "TMe-3200"
#>
#> $III
#> [1] "TMe-617" "TMe-2502" "TMe-161" "TMe-2356" "TMe-70" "TMe-2394" "TMe-3335"
#> [8] "TMe-2968" "TMe-1790"
#>
#> $IV
#> [1] "TMe-1139" "TMe-12" "TMe-3409" "TMe-1020" "TMe-1027" "TMe-170"
#> [7] "TMe-525" "TMe-3562" "TMe-3255" "TMe-761" "TMe-2567" "TMe-3273"
#> [13] "TMe-3581" "TMe-2039" "TMe-1988" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-332" "TMe-344" "TMe-1414" "TMe-2750" "TMe-532" "TMe-1220"
#> [7] "TMe-167" "TMe-439" "TMe-256" "TMe-487" "TMe-723" "TMe-1880"
#> [13] "TMe-667" "TMe-2853" "TMe-3329" "TMe-712" "TMe-2355" "TMe-816"
#> [19] "TMe-2003" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1608" "TMe-2196" "TMe-936" "TMe-1217" "TMe-620" "TMe-3177"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out1,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "average")
# Cluster-Based Sampling via Hierarchical Clustering with Medoid Selection
## UPGMA
sel_hclust_medoid_out1 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "average")
sel_hclust_medoid_out1
#> $I
#> [1] "TMe-1823" "TMe-3132" "TMe-44" "TMe-3539" "TMe-952" "TMe-500"
#> [7] "TMe-3110" "TMe-815" "TMe-2934" "TMe-566" "TMe-2967" "TMe-3398"
#> [13] "TMe-1425" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2258" "TMe-681" "TMe-890" "TMe-2951" "TMe-3557" "TMe-3239"
#> [7] "TMe-674" "TMe-2352" "TMe-1831" "TMe-3101" "TMe-2033" "TMe-2952"
#> [13] "TMe-3284" "TMe-171" "TMe-3200"
#>
#> $III
#> [1] "TMe-617" "TMe-2502" "TMe-161" "TMe-2356" "TMe-70" "TMe-2394" "TMe-3335"
#> [8] "TMe-2968" "TMe-1790"
#>
#> $IV
#> [1] "TMe-1139" "TMe-12" "TMe-3409" "TMe-1020" "TMe-1027" "TMe-170"
#> [7] "TMe-525" "TMe-3562" "TMe-3255" "TMe-761" "TMe-2567" "TMe-3273"
#> [13] "TMe-3581" "TMe-2039" "TMe-1988" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-332" "TMe-344" "TMe-1414" "TMe-2750" "TMe-532" "TMe-1220"
#> [7] "TMe-167" "TMe-439" "TMe-256" "TMe-487" "TMe-723" "TMe-1880"
#> [13] "TMe-667" "TMe-2853" "TMe-3329" "TMe-712" "TMe-2355" "TMe-816"
#> [19] "TMe-2003" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1608" "TMe-2196" "TMe-936" "TMe-1217" "TMe-620" "TMe-3177"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out1,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "average")
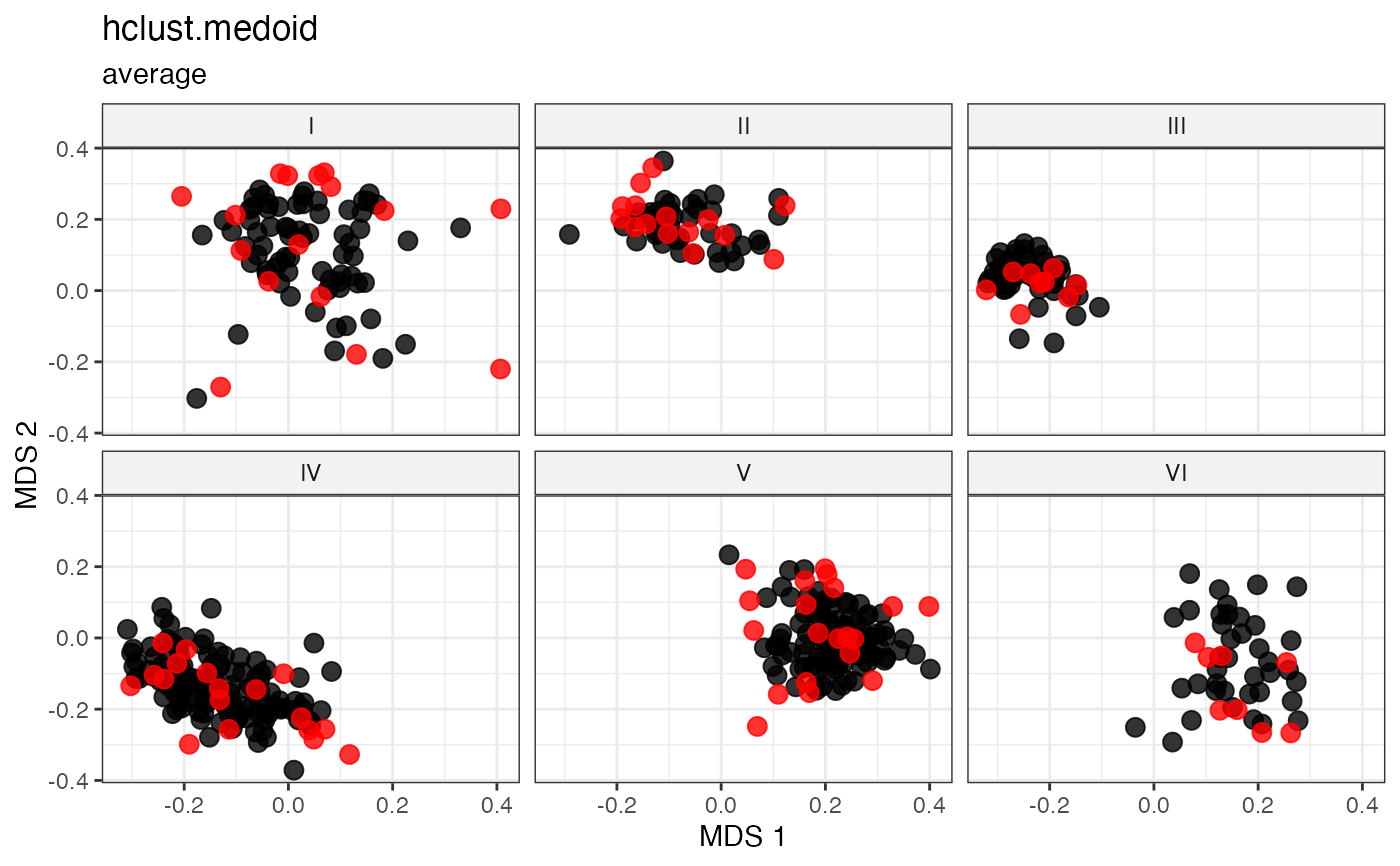 ## Single-linkage
sel_hclust_medoid_out2 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "single")
sel_hclust_medoid_out2
#> $I
#> [1] "TMe-1823" "TMe-3051" "TMe-815" "TMe-2934" "TMe-566" "TMe-3481"
#> [7] "TMe-2967" "TMe-3398" "TMe-1425" "TMe-569" "TMe-3087" "TMe-3112"
#> [13] "TMe-2785" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2021" "TMe-455" "TMe-674" "TMe-2352" "TMe-2568" "TMe-1668"
#> [7] "TMe-369" "TMe-2033" "TMe-2952" "TMe-3093" "TMe-40" "TMe-3805"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-2748" "TMe-2394" "TMe-234" "TMe-2086" "TMe-381" "TMe-3631" "TMe-261"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-428" "TMe-2956" "TMe-1020" "TMe-3390" "TMe-3255" "TMe-761"
#> [7] "TMe-2567" "TMe-3527" "TMe-3273" "TMe-372" "TMe-3189" "TMe-698"
#> [13] "TMe-1988" "TMe-186" "TMe-812" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-332" "TMe-1375" "TMe-344" "TMe-1534" "TMe-487" "TMe-723"
#> [7] "TMe-2290" "TMe-667" "TMe-2853" "TMe-3329" "TMe-712" "TMe-362"
#> [13] "TMe-2355" "TMe-419" "TMe-2003" "TMe-730" "TMe-98" "TMe-2907"
#> [19] "TMe-2425" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-620" "TMe-751" "TMe-1646" "TMe-1124" "TMe-310" "TMe-598"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out2,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "single")
## Single-linkage
sel_hclust_medoid_out2 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "single")
sel_hclust_medoid_out2
#> $I
#> [1] "TMe-1823" "TMe-3051" "TMe-815" "TMe-2934" "TMe-566" "TMe-3481"
#> [7] "TMe-2967" "TMe-3398" "TMe-1425" "TMe-569" "TMe-3087" "TMe-3112"
#> [13] "TMe-2785" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2021" "TMe-455" "TMe-674" "TMe-2352" "TMe-2568" "TMe-1668"
#> [7] "TMe-369" "TMe-2033" "TMe-2952" "TMe-3093" "TMe-40" "TMe-3805"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-2748" "TMe-2394" "TMe-234" "TMe-2086" "TMe-381" "TMe-3631" "TMe-261"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-428" "TMe-2956" "TMe-1020" "TMe-3390" "TMe-3255" "TMe-761"
#> [7] "TMe-2567" "TMe-3527" "TMe-3273" "TMe-372" "TMe-3189" "TMe-698"
#> [13] "TMe-1988" "TMe-186" "TMe-812" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-332" "TMe-1375" "TMe-344" "TMe-1534" "TMe-487" "TMe-723"
#> [7] "TMe-2290" "TMe-667" "TMe-2853" "TMe-3329" "TMe-712" "TMe-362"
#> [13] "TMe-2355" "TMe-419" "TMe-2003" "TMe-730" "TMe-98" "TMe-2907"
#> [19] "TMe-2425" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-620" "TMe-751" "TMe-1646" "TMe-1124" "TMe-310" "TMe-598"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out2,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "single")
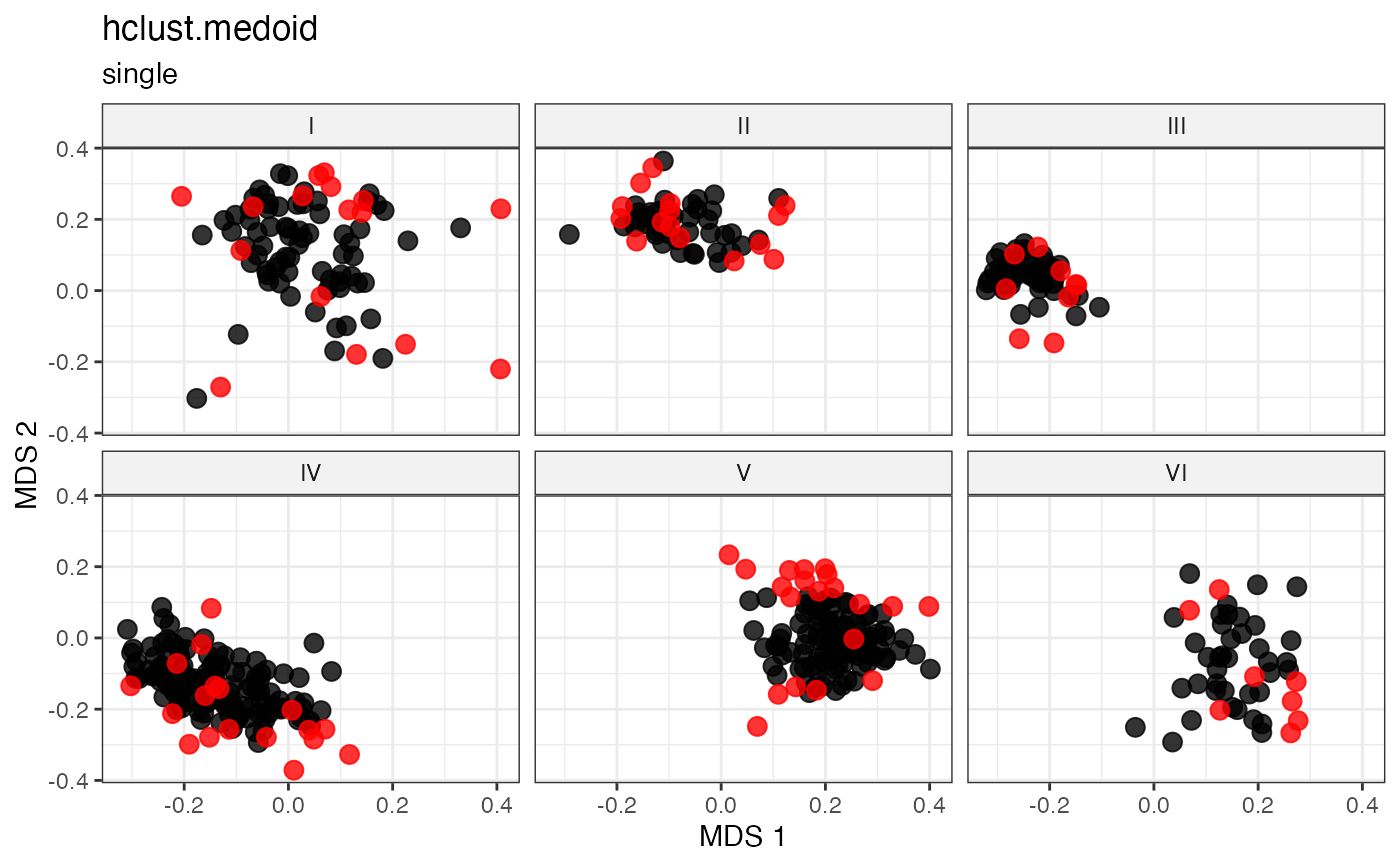 ## Complete-linkage
sel_hclust_medoid_out3 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "complete")
sel_hclust_medoid_out3
#> $I
#> [1] "TMe-2103" "TMe-28" "TMe-44" "TMe-3539" "TMe-1823" "TMe-1117"
#> [7] "TMe-500" "TMe-1170" "TMe-2944" "TMe-3623" "TMe-3115" "TMe-566"
#> [13] "TMe-3130" "TMe-1218" "TMe-1425" "TMe-1830"
#>
#> $II
#> [1] "TMe-2258" "TMe-681" "TMe-3800" "TMe-2951" "TMe-3557" "TMe-3239"
#> [7] "TMe-2995" "TMe-1385" "TMe-2352" "TMe-1831" "TMe-1668" "TMe-409"
#> [13] "TMe-2033" "TMe-2952" "TMe-3284"
#>
#> $III
#> [1] "TMe-3207" "TMe-2270" "TMe-161" "TMe-2356" "TMe-2394" "TMe-3335" "TMe-1738"
#> [8] "TMe-2733" "TMe-1790"
#>
#> $IV
#> [1] "TMe-3269" "TMe-2947" "TMe-1139" "TMe-3585" "TMe-1155" "TMe-1336"
#> [7] "TMe-3378" "TMe-3758" "TMe-1027" "TMe-57" "TMe-2458" "TMe-525"
#> [13] "TMe-3327" "TMe-761" "TMe-2928" "TMe-3581" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-574" "TMe-629" "TMe-892" "TMe-363" "TMe-2124" "TMe-2750"
#> [7] "TMe-532" "TMe-1220" "TMe-1160" "TMe-954" "TMe-1622" "TMe-647"
#> [13] "TMe-288" "TMe-256" "TMe-1234" "TMe-423" "TMe-2853" "TMe-3329"
#> [19] "TMe-623" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1744" "TMe-1062" "TMe-968" "TMe-1445" "TMe-620" "TMe-3177"
#> [8] "TMe-1035"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out3,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "complete")
## Complete-linkage
sel_hclust_medoid_out3 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "complete")
sel_hclust_medoid_out3
#> $I
#> [1] "TMe-2103" "TMe-28" "TMe-44" "TMe-3539" "TMe-1823" "TMe-1117"
#> [7] "TMe-500" "TMe-1170" "TMe-2944" "TMe-3623" "TMe-3115" "TMe-566"
#> [13] "TMe-3130" "TMe-1218" "TMe-1425" "TMe-1830"
#>
#> $II
#> [1] "TMe-2258" "TMe-681" "TMe-3800" "TMe-2951" "TMe-3557" "TMe-3239"
#> [7] "TMe-2995" "TMe-1385" "TMe-2352" "TMe-1831" "TMe-1668" "TMe-409"
#> [13] "TMe-2033" "TMe-2952" "TMe-3284"
#>
#> $III
#> [1] "TMe-3207" "TMe-2270" "TMe-161" "TMe-2356" "TMe-2394" "TMe-3335" "TMe-1738"
#> [8] "TMe-2733" "TMe-1790"
#>
#> $IV
#> [1] "TMe-3269" "TMe-2947" "TMe-1139" "TMe-3585" "TMe-1155" "TMe-1336"
#> [7] "TMe-3378" "TMe-3758" "TMe-1027" "TMe-57" "TMe-2458" "TMe-525"
#> [13] "TMe-3327" "TMe-761" "TMe-2928" "TMe-3581" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-574" "TMe-629" "TMe-892" "TMe-363" "TMe-2124" "TMe-2750"
#> [7] "TMe-532" "TMe-1220" "TMe-1160" "TMe-954" "TMe-1622" "TMe-647"
#> [13] "TMe-288" "TMe-256" "TMe-1234" "TMe-423" "TMe-2853" "TMe-3329"
#> [19] "TMe-623" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1744" "TMe-1062" "TMe-968" "TMe-1445" "TMe-620" "TMe-3177"
#> [8] "TMe-1035"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out3,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "complete")
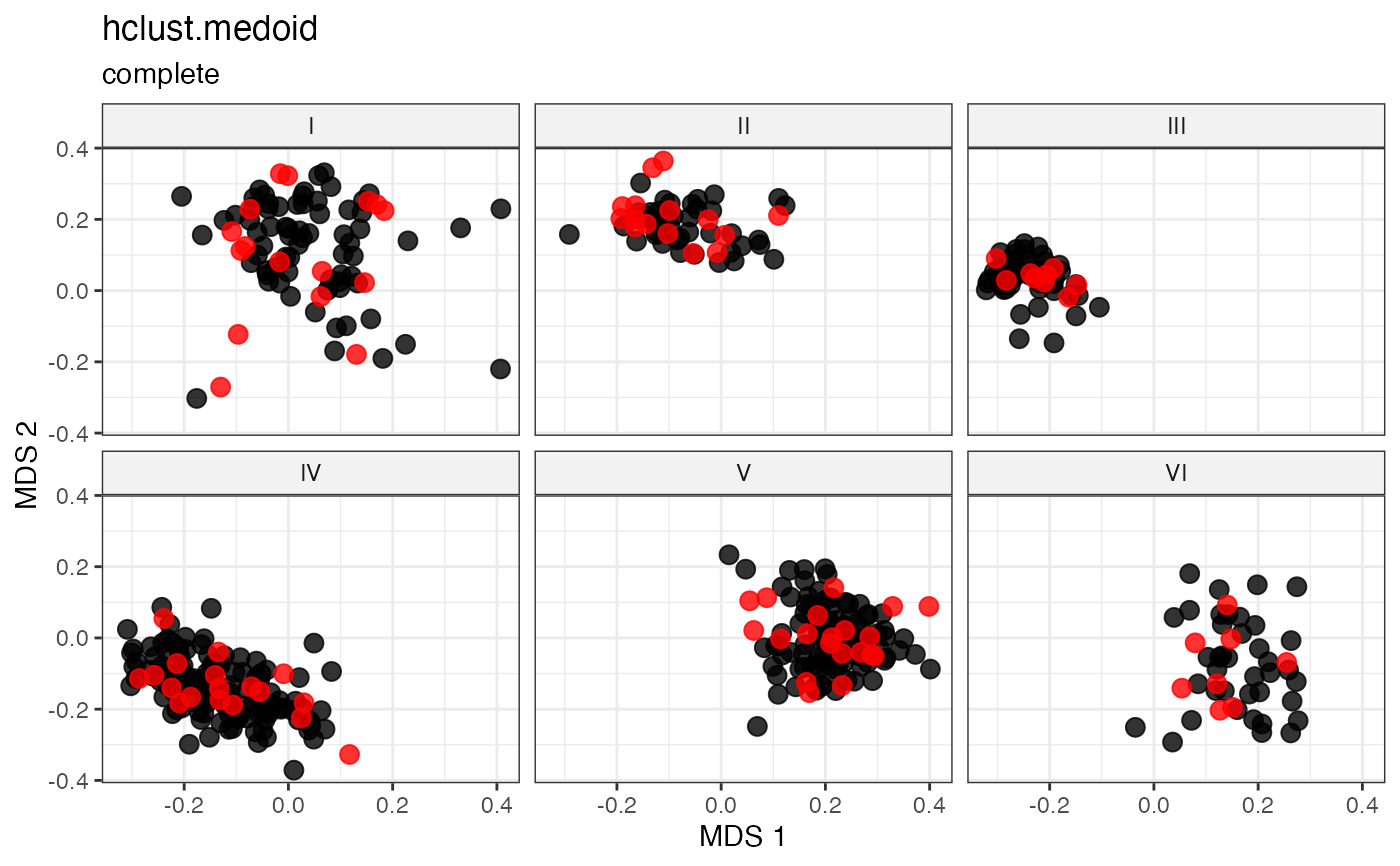 ## Ward's D
sel_hclust_medoid_out4 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "ward.D")
sel_hclust_medoid_out4
#> $I
#> [1] "TMe-1717" "TMe-3481" "TMe-44" "TMe-3539" "TMe-1823" "TMe-952"
#> [7] "TMe-500" "TMe-865" "TMe-1532" "TMe-2237" "TMe-2964" "TMe-566"
#> [13] "TMe-882" "TMe-28" "TMe-3115" "TMe-1830"
#>
#> $II
#> [1] "TMe-2021" "TMe-2715" "TMe-890" "TMe-2951" "TMe-3557" "TMe-3239"
#> [7] "TMe-2995" "TMe-674" "TMe-3805" "TMe-1831" "TMe-1754" "TMe-1668"
#> [13] "TMe-409" "TMe-1474" "TMe-3284"
#>
#> $III
#> [1] "TMe-3007" "TMe-2502" "TMe-161" "TMe-2356" "TMe-70" "TMe-1230" "TMe-3335"
#> [8] "TMe-3569" "TMe-1790"
#>
#> $IV
#> [1] "TMe-3103" "TMe-3558" "TMe-1144" "TMe-3409" "TMe-1377" "TMe-1336"
#> [7] "TMe-3265" "TMe-1996" "TMe-622" "TMe-1139" "TMe-3562" "TMe-57"
#> [13] "TMe-63" "TMe-525" "TMe-3068" "TMe-1027" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1979" "TMe-629" "TMe-892" "TMe-1234" "TMe-1427" "TMe-406"
#> [7] "TMe-1788" "TMe-1160" "TMe-954" "TMe-439" "TMe-247" "TMe-645"
#> [13] "TMe-256" "TMe-730" "TMe-1622" "TMe-1760" "TMe-667" "TMe-2853"
#> [19] "TMe-307" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1608" "TMe-2196" "TMe-531" "TMe-936" "TMe-1217" "TMe-620"
#> [8] "TMe-1445"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out4,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "ward.D")
## Ward's D
sel_hclust_medoid_out4 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "ward.D")
sel_hclust_medoid_out4
#> $I
#> [1] "TMe-1717" "TMe-3481" "TMe-44" "TMe-3539" "TMe-1823" "TMe-952"
#> [7] "TMe-500" "TMe-865" "TMe-1532" "TMe-2237" "TMe-2964" "TMe-566"
#> [13] "TMe-882" "TMe-28" "TMe-3115" "TMe-1830"
#>
#> $II
#> [1] "TMe-2021" "TMe-2715" "TMe-890" "TMe-2951" "TMe-3557" "TMe-3239"
#> [7] "TMe-2995" "TMe-674" "TMe-3805" "TMe-1831" "TMe-1754" "TMe-1668"
#> [13] "TMe-409" "TMe-1474" "TMe-3284"
#>
#> $III
#> [1] "TMe-3007" "TMe-2502" "TMe-161" "TMe-2356" "TMe-70" "TMe-1230" "TMe-3335"
#> [8] "TMe-3569" "TMe-1790"
#>
#> $IV
#> [1] "TMe-3103" "TMe-3558" "TMe-1144" "TMe-3409" "TMe-1377" "TMe-1336"
#> [7] "TMe-3265" "TMe-1996" "TMe-622" "TMe-1139" "TMe-3562" "TMe-57"
#> [13] "TMe-63" "TMe-525" "TMe-3068" "TMe-1027" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1979" "TMe-629" "TMe-892" "TMe-1234" "TMe-1427" "TMe-406"
#> [7] "TMe-1788" "TMe-1160" "TMe-954" "TMe-439" "TMe-247" "TMe-645"
#> [13] "TMe-256" "TMe-730" "TMe-1622" "TMe-1760" "TMe-667" "TMe-2853"
#> [19] "TMe-307" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1608" "TMe-2196" "TMe-531" "TMe-936" "TMe-1217" "TMe-620"
#> [8] "TMe-1445"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out4,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "ward.D")
 ## WPGMA
sel_hclust_medoid_out5 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "mcquitty")
sel_hclust_medoid_out5
#> $I
#> [1] "TMe-1717" "TMe-3132" "TMe-1170" "TMe-3539" "TMe-952" "TMe-500"
#> [7] "TMe-2944" "TMe-2943" "TMe-815" "TMe-566" "TMe-41" "TMe-2967"
#> [13] "TMe-1425" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2258" "TMe-2715" "TMe-3800" "TMe-2951" "TMe-3557" "TMe-3101"
#> [7] "TMe-674" "TMe-2352" "TMe-1698" "TMe-1668" "TMe-409" "TMe-2033"
#> [13] "TMe-2952" "TMe-171" "TMe-3200"
#>
#> $III
#> [1] "TMe-617" "TMe-2394" "TMe-161" "TMe-2356" "TMe-70" "TMe-2086" "TMe-3335"
#> [8] "TMe-2968" "TMe-1790"
#>
#> $IV
#> [1] "TMe-428" "TMe-1167" "TMe-1144" "TMe-3409" "TMe-1020" "TMe-353"
#> [7] "TMe-170" "TMe-2946" "TMe-3255" "TMe-761" "TMe-2567" "TMe-3273"
#> [13] "TMe-3581" "TMe-2924" "TMe-1988" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1979" "TMe-629" "TMe-344" "TMe-745" "TMe-2750" "TMe-532"
#> [7] "TMe-1788" "TMe-167" "TMe-247" "TMe-647" "TMe-256" "TMe-723"
#> [13] "TMe-1880" "TMe-1401" "TMe-419" "TMe-2853" "TMe-3329" "TMe-2355"
#> [19] "TMe-2003" "TMe-2018"
#>
#> $VI
#> [1] "TMe-936" "TMe-1744" "TMe-2196" "TMe-1302" "TMe-1217" "TMe-1445" "TMe-3177"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out5,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "mcquitty")
## WPGMA
sel_hclust_medoid_out5 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "mcquitty")
sel_hclust_medoid_out5
#> $I
#> [1] "TMe-1717" "TMe-3132" "TMe-1170" "TMe-3539" "TMe-952" "TMe-500"
#> [7] "TMe-2944" "TMe-2943" "TMe-815" "TMe-566" "TMe-41" "TMe-2967"
#> [13] "TMe-1425" "TMe-3437" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-2258" "TMe-2715" "TMe-3800" "TMe-2951" "TMe-3557" "TMe-3101"
#> [7] "TMe-674" "TMe-2352" "TMe-1698" "TMe-1668" "TMe-409" "TMe-2033"
#> [13] "TMe-2952" "TMe-171" "TMe-3200"
#>
#> $III
#> [1] "TMe-617" "TMe-2394" "TMe-161" "TMe-2356" "TMe-70" "TMe-2086" "TMe-3335"
#> [8] "TMe-2968" "TMe-1790"
#>
#> $IV
#> [1] "TMe-428" "TMe-1167" "TMe-1144" "TMe-3409" "TMe-1020" "TMe-353"
#> [7] "TMe-170" "TMe-2946" "TMe-3255" "TMe-761" "TMe-2567" "TMe-3273"
#> [13] "TMe-3581" "TMe-2924" "TMe-1988" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1979" "TMe-629" "TMe-344" "TMe-745" "TMe-2750" "TMe-532"
#> [7] "TMe-1788" "TMe-167" "TMe-247" "TMe-647" "TMe-256" "TMe-723"
#> [13] "TMe-1880" "TMe-1401" "TMe-419" "TMe-2853" "TMe-3329" "TMe-2355"
#> [19] "TMe-2003" "TMe-2018"
#>
#> $VI
#> [1] "TMe-936" "TMe-1744" "TMe-2196" "TMe-1302" "TMe-1217" "TMe-1445" "TMe-3177"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out5,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "mcquitty")
 ## WPGMC
sel_hclust_medoid_out6 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "median")
sel_hclust_medoid_out6
#> $I
#> [1] "TMe-1823" "TMe-3051" "TMe-815" "TMe-2934" "TMe-3262" "TMe-2943"
#> [7] "TMe-2967" "TMe-1096" "TMe-1425" "TMe-569" "TMe-3112" "TMe-2785"
#> [13] "TMe-3437" "TMe-3111" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-3495" "TMe-3800" "TMe-674" "TMe-2352" "TMe-3766" "TMe-2329"
#> [7] "TMe-369" "TMe-2033" "TMe-2952" "TMe-3093" "TMe-3805" "TMe-3547"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-1787" "TMe-1725" "TMe-3100" "TMe-3631" "TMe-14" "TMe-261" "TMe-3397"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-428" "TMe-2956" "TMe-1020" "TMe-1376" "TMe-3255" "TMe-761"
#> [7] "TMe-3527" "TMe-3273" "TMe-3189" "TMe-698" "TMe-1434" "TMe-427"
#> [13] "TMe-2971" "TMe-1988" "TMe-186" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-332" "TMe-344" "TMe-769" "TMe-2151" "TMe-256" "TMe-723"
#> [7] "TMe-730" "TMe-2290" "TMe-2853" "TMe-3329" "TMe-423" "TMe-712"
#> [13] "TMe-362" "TMe-2355" "TMe-419" "TMe-1934" "TMe-2003" "TMe-2425"
#> [19] "TMe-2855" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-693" "TMe-751" "TMe-1646" "TMe-1124" "TMe-1661" "TMe-2791"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out6,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "median")
## WPGMC
sel_hclust_medoid_out6 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "median")
sel_hclust_medoid_out6
#> $I
#> [1] "TMe-1823" "TMe-3051" "TMe-815" "TMe-2934" "TMe-3262" "TMe-2943"
#> [7] "TMe-2967" "TMe-1096" "TMe-1425" "TMe-569" "TMe-3112" "TMe-2785"
#> [13] "TMe-3437" "TMe-3111" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-3495" "TMe-3800" "TMe-674" "TMe-2352" "TMe-3766" "TMe-2329"
#> [7] "TMe-369" "TMe-2033" "TMe-2952" "TMe-3093" "TMe-3805" "TMe-3547"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-1787" "TMe-1725" "TMe-3100" "TMe-3631" "TMe-14" "TMe-261" "TMe-3397"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-428" "TMe-2956" "TMe-1020" "TMe-1376" "TMe-3255" "TMe-761"
#> [7] "TMe-3527" "TMe-3273" "TMe-3189" "TMe-698" "TMe-1434" "TMe-427"
#> [13] "TMe-2971" "TMe-1988" "TMe-186" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-332" "TMe-344" "TMe-769" "TMe-2151" "TMe-256" "TMe-723"
#> [7] "TMe-730" "TMe-2290" "TMe-2853" "TMe-3329" "TMe-423" "TMe-712"
#> [13] "TMe-362" "TMe-2355" "TMe-419" "TMe-1934" "TMe-2003" "TMe-2425"
#> [19] "TMe-2855" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-693" "TMe-751" "TMe-1646" "TMe-1124" "TMe-1661" "TMe-2791"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out6,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "median")
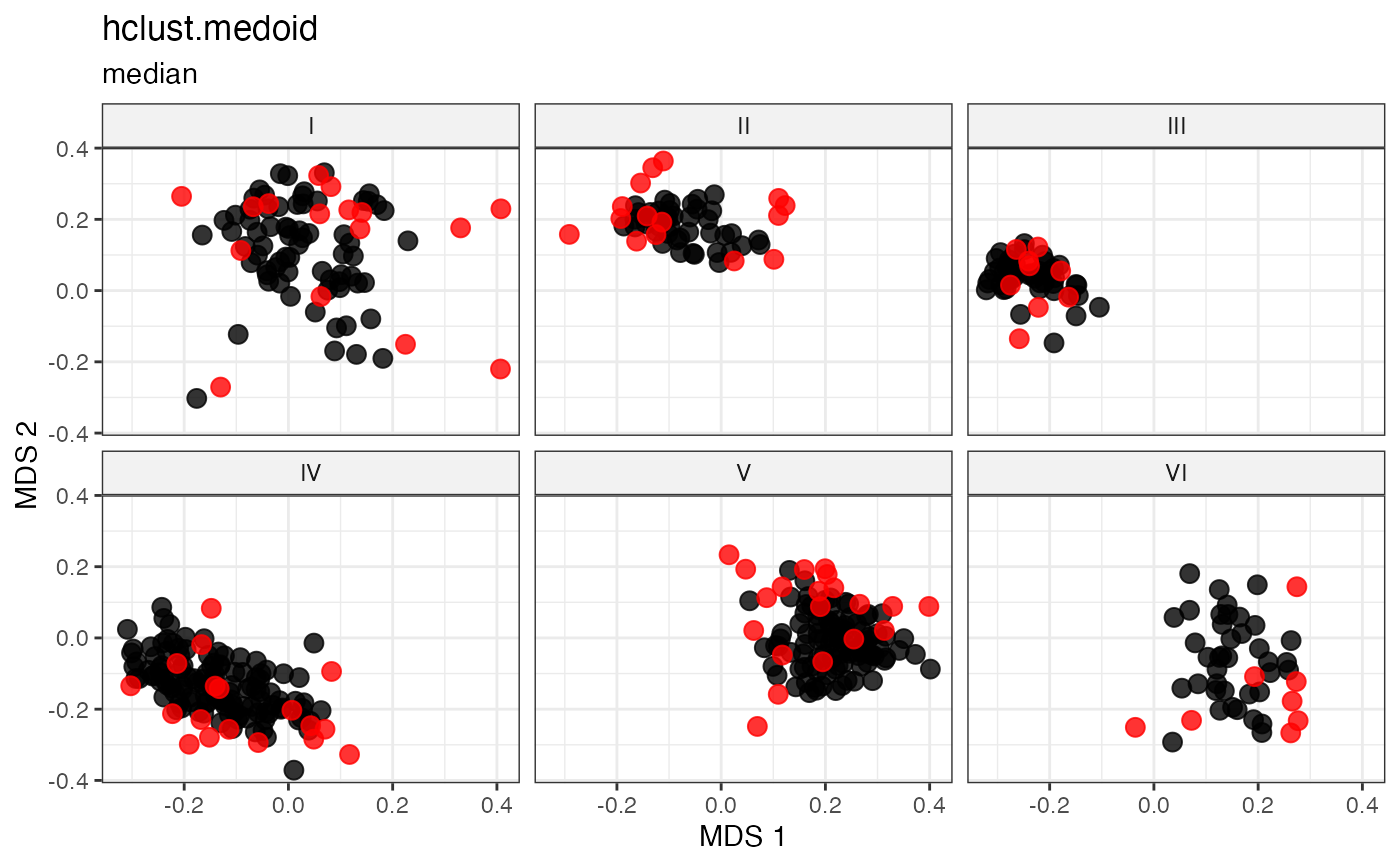 ## UPGMC
sel_hclust_medoid_out7 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "centroid")
sel_hclust_medoid_out7
#> $I
#> [1] "TMe-1823" "TMe-500" "TMe-815" "TMe-2934" "TMe-566" "TMe-2943"
#> [7] "TMe-1218" "TMe-2967" "TMe-1190" "TMe-1425" "TMe-569" "TMe-41"
#> [13] "TMe-3437" "TMe-2027" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-3495" "TMe-3800" "TMe-674" "TMe-2352" "TMe-2568" "TMe-3766"
#> [7] "TMe-2329" "TMe-369" "TMe-2033" "TMe-2952" "TMe-1754" "TMe-3805"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-1725" "TMe-234" "TMe-381" "TMe-3631" "TMe-2374" "TMe-3715" "TMe-261"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-428" "TMe-2956" "TMe-1020" "TMe-1376" "TMe-761" "TMe-2567"
#> [7] "TMe-3527" "TMe-3273" "TMe-3581" "TMe-27" "TMe-698" "TMe-1434"
#> [13] "TMe-427" "TMe-1988" "TMe-812" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-332" "TMe-1375" "TMe-344" "TMe-532" "TMe-723" "TMe-2290"
#> [7] "TMe-667" "TMe-224" "TMe-2853" "TMe-3329" "TMe-423" "TMe-712"
#> [13] "TMe-2355" "TMe-419" "TMe-2003" "TMe-627" "TMe-98" "TMe-2750"
#> [19] "TMe-2907" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-693" "TMe-751" "TMe-1124" "TMe-2791" "TMe-1035" "TMe-1992"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out7,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "centroid")
## UPGMC
sel_hclust_medoid_out7 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "centroid")
sel_hclust_medoid_out7
#> $I
#> [1] "TMe-1823" "TMe-500" "TMe-815" "TMe-2934" "TMe-566" "TMe-2943"
#> [7] "TMe-1218" "TMe-2967" "TMe-1190" "TMe-1425" "TMe-569" "TMe-41"
#> [13] "TMe-3437" "TMe-2027" "TMe-3685" "TMe-1830"
#>
#> $II
#> [1] "TMe-3495" "TMe-3800" "TMe-674" "TMe-2352" "TMe-2568" "TMe-3766"
#> [7] "TMe-2329" "TMe-369" "TMe-2033" "TMe-2952" "TMe-1754" "TMe-3805"
#> [13] "TMe-171" "TMe-1385" "TMe-3200"
#>
#> $III
#> [1] "TMe-1725" "TMe-234" "TMe-381" "TMe-3631" "TMe-2374" "TMe-3715" "TMe-261"
#> [8] "TMe-425" "TMe-1790"
#>
#> $IV
#> [1] "TMe-428" "TMe-2956" "TMe-1020" "TMe-1376" "TMe-761" "TMe-2567"
#> [7] "TMe-3527" "TMe-3273" "TMe-3581" "TMe-27" "TMe-698" "TMe-1434"
#> [13] "TMe-427" "TMe-1988" "TMe-812" "TMe-241" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-332" "TMe-1375" "TMe-344" "TMe-532" "TMe-723" "TMe-2290"
#> [7] "TMe-667" "TMe-224" "TMe-2853" "TMe-3329" "TMe-423" "TMe-712"
#> [13] "TMe-2355" "TMe-419" "TMe-2003" "TMe-627" "TMe-98" "TMe-2750"
#> [19] "TMe-2907" "TMe-2018"
#>
#> $VI
#> [1] "TMe-809" "TMe-693" "TMe-751" "TMe-1124" "TMe-2791" "TMe-1035" "TMe-1992"
#> [8] "TMe-2983"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out7,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "centroid")
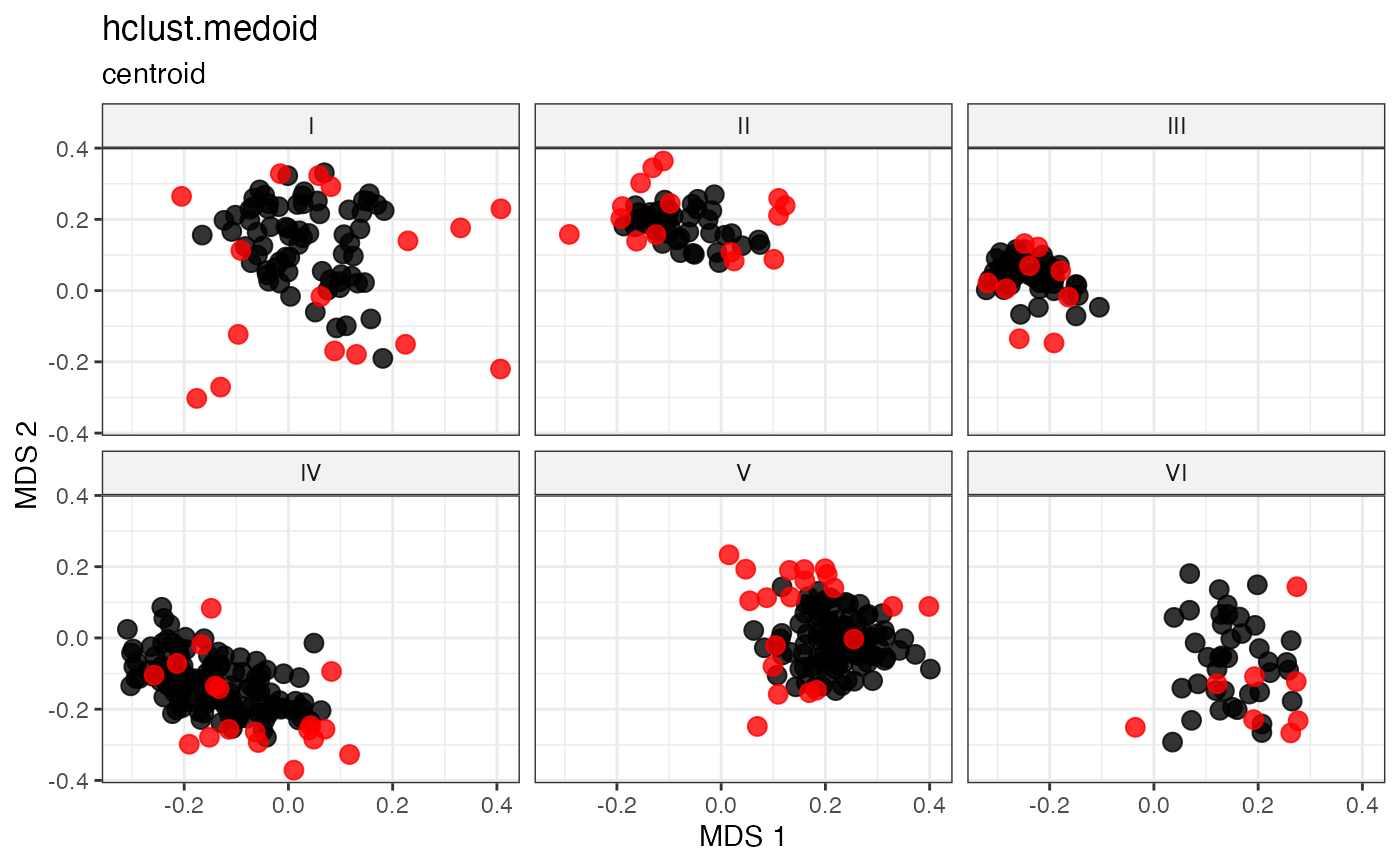 ## Ward's D2
sel_hclust_medoid_out8 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "ward.D2")
sel_hclust_medoid_out8
#> $I
#> [1] "TMe-1717" "TMe-3481" "TMe-500" "TMe-44" "TMe-3539" "TMe-1823"
#> [7] "TMe-952" "TMe-882" "TMe-3110" "TMe-2237" "TMe-2964" "TMe-566"
#> [13] "TMe-3130" "TMe-28" "TMe-3115" "TMe-1830"
#>
#> $II
#> [1] "TMe-2021" "TMe-2715" "TMe-890" "TMe-2951" "TMe-3557" "TMe-3239"
#> [7] "TMe-2995" "TMe-674" "TMe-2352" "TMe-1831" "TMe-1754" "TMe-1668"
#> [13] "TMe-1474" "TMe-2033" "TMe-3284"
#>
#> $III
#> [1] "TMe-3007" "TMe-2502" "TMe-161" "TMe-2356" "TMe-70" "TMe-1230" "TMe-3335"
#> [8] "TMe-3569" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2318" "TMe-3558" "TMe-1144" "TMe-2958" "TMe-1377" "TMe-1336"
#> [7] "TMe-3265" "TMe-387" "TMe-1139" "TMe-3562" "TMe-1456" "TMe-2947"
#> [13] "TMe-3068" "TMe-428" "TMe-525" "TMe-1027" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1979" "TMe-629" "TMe-892" "TMe-1234" "TMe-1427" "TMe-406"
#> [7] "TMe-1788" "TMe-1160" "TMe-954" "TMe-439" "TMe-247" "TMe-645"
#> [13] "TMe-256" "TMe-730" "TMe-1622" "TMe-1760" "TMe-667" "TMe-2853"
#> [19] "TMe-3329" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1608" "TMe-2196" "TMe-222" "TMe-1445" "TMe-936" "TMe-1217"
#> [8] "TMe-620"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out8,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "ward.D2")
## Ward's D2
sel_hclust_medoid_out8 <-
select.distance(data = data, names = "genotypes",
group = "Cluster", alloc = counts,
dist.mat = dist_matrix,
always.selected = mand_accns,
method = "hclust.medoid",
hclust.method = "ward.D2")
sel_hclust_medoid_out8
#> $I
#> [1] "TMe-1717" "TMe-3481" "TMe-500" "TMe-44" "TMe-3539" "TMe-1823"
#> [7] "TMe-952" "TMe-882" "TMe-3110" "TMe-2237" "TMe-2964" "TMe-566"
#> [13] "TMe-3130" "TMe-28" "TMe-3115" "TMe-1830"
#>
#> $II
#> [1] "TMe-2021" "TMe-2715" "TMe-890" "TMe-2951" "TMe-3557" "TMe-3239"
#> [7] "TMe-2995" "TMe-674" "TMe-2352" "TMe-1831" "TMe-1754" "TMe-1668"
#> [13] "TMe-1474" "TMe-2033" "TMe-3284"
#>
#> $III
#> [1] "TMe-3007" "TMe-2502" "TMe-161" "TMe-2356" "TMe-70" "TMe-1230" "TMe-3335"
#> [8] "TMe-3569" "TMe-1790"
#>
#> $IV
#> [1] "TMe-2318" "TMe-3558" "TMe-1144" "TMe-2958" "TMe-1377" "TMe-1336"
#> [7] "TMe-3265" "TMe-387" "TMe-1139" "TMe-3562" "TMe-1456" "TMe-2947"
#> [13] "TMe-3068" "TMe-428" "TMe-525" "TMe-1027" "TMe-801" "TMe-3191"
#>
#> $V
#> [1] "TMe-1979" "TMe-629" "TMe-892" "TMe-1234" "TMe-1427" "TMe-406"
#> [7] "TMe-1788" "TMe-1160" "TMe-954" "TMe-439" "TMe-247" "TMe-645"
#> [13] "TMe-256" "TMe-730" "TMe-1622" "TMe-1760" "TMe-667" "TMe-2853"
#> [19] "TMe-3329" "TMe-2018"
#>
#> $VI
#> [1] "TMe-1481" "TMe-1608" "TMe-2196" "TMe-222" "TMe-1445" "TMe-936" "TMe-1217"
#> [8] "TMe-620"
#>
plot_dist(d = dist_matrix, method = "isomds",
gp = gp_vec,
highlight = unlist(sel_hclust_medoid_out8,
use.names = FALSE)) +
labs(title = "hclust.medoid", subtitle = "ward.D2")
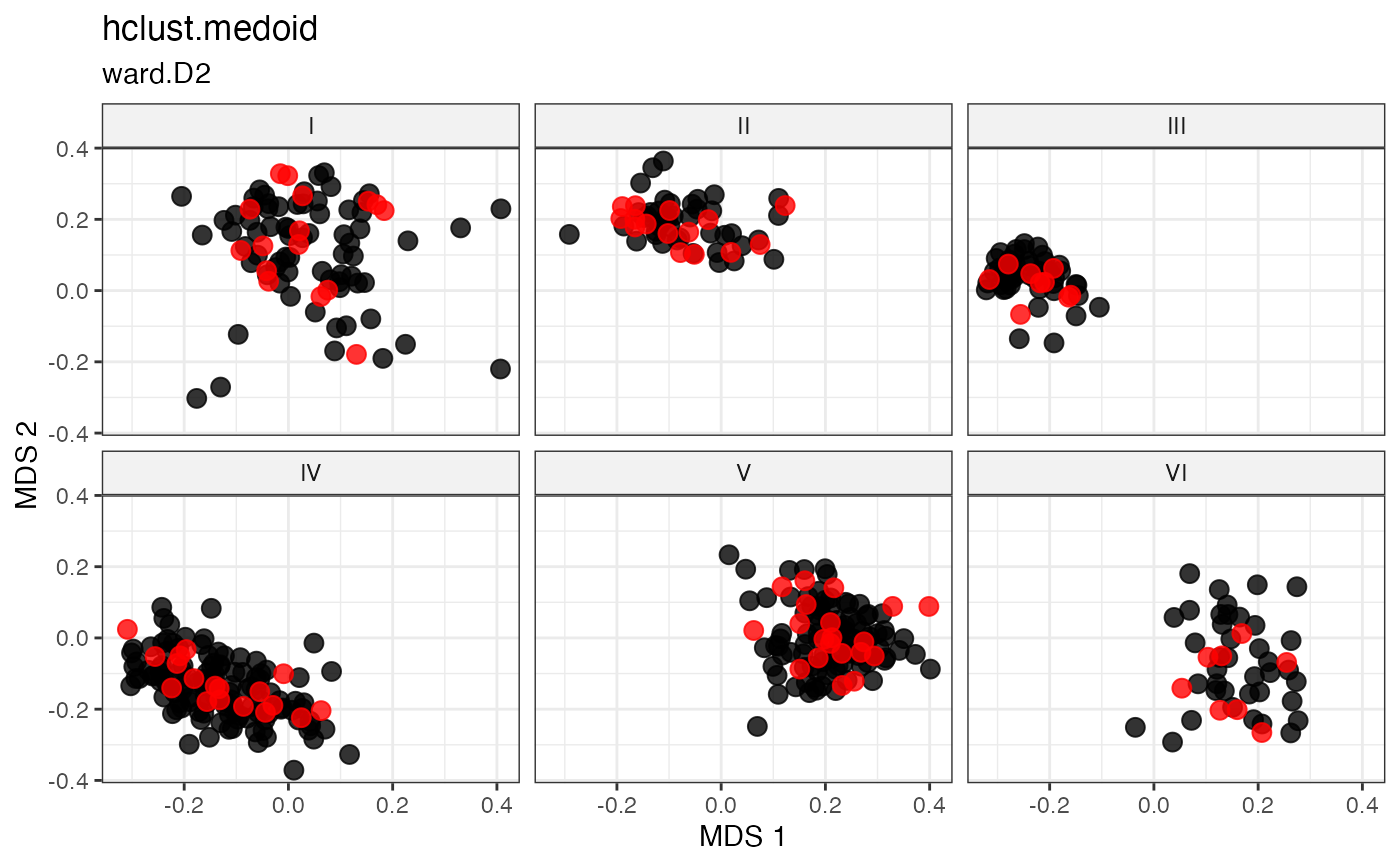 # }
# }